Descargar la presentación
La descarga está en progreso. Por favor, espere

1
Reunión clínica Neurología: Convulsiones neonatales Status Epileptico Neonatal Microlisencefalia
Dr. Fernando Subercaseaux Becado neonatología U Chile Rotación Neurología Infantil Abril 2014

2
Caso clínico G.J.A. 1 Antecedentes prenatales:
Madre M2. Padres no consanguíneos Mal control embarazo (particular). Consumo marihuana y alcohol durante embarazo. Además de medicamentos naturales “adelgazantes” Solo registro eco previas a las 29+1 y 31+6 semanas Eco urgencia: RCIU- SFA (PBF): 22/3 Obs. sufrimiento fetal agudo, LA normal, doppler normal, anatomía normal, EPF 2140 gramos, 37 semanas RCIU <p3 Se indica cesárea urgencia
. Consumo marihuana y alcohol durante embarazo. Además de medicamentos naturales adelgazantes Solo registro eco previas a las 29+1 y 31+6 semanas. Eco urgencia: RCIU- SFA (PBF): 22/3 Obs. sufrimiento fetal agudo, LA normal, doppler normal, anatomía normal, EPF 2140 gramos, 37 semanas. RCIU <p3. Se indica cesárea urgencia.")
3
Caso clínico G.J.A. 2 Antecedente parto:
Fecha nacimiento 22/3/ :14 horas Nace deprimido, requiere ventilación a presión positiva. Peso nacimiento 2100, talla nacimiento: 44, CC 29, edad gestacional 37 semanas. Sexo masculino. Apgar 7-8 Gases cordón: pH 7,4, po2 25, pco2 35, be -2

4
G.J.A RNT PEG 22/3/2014 – 329 Apgar: 7-8 SEGUNDO HIJO Padres sanos No consanguíneos Crisis clónicas 4 extrem 5-10 segundos Crisis igual características Responde a terapia AC 2 horas 24 horas 36 horas ingreso UCI NEO: microcefalia < p3. hipospadia-? VM Examen: Sigue en vm, poco reactivos Tac cerebral: micro- Lisencefalia con hipoplasia Cerebelar (Dr. camelio) Glicemia - amonio función renal GSA – ELP, ca-p- Screening Infección Ecoscopia cerebral no concluyente Re control metabólico Básico normal EEG: severamente Alterado, actividad Epileptiforme centro Temporal bilateral 4 crisis con clínica Carga Fenitoína 15 mg/k Midazolam bic 0,1 Piridoxina 100 mg/d Midazolam 0,2 mg/k Fenobarbital + mdz
 Glicemia - amonio. función renal. GSA – ELP, ca-p- Screening Infección. Ecoscopia cerebral no. concluyente. Re control metabólico. Básico normal. EEG: severamente. Alterado, actividad. Epileptiforme centro. Temporal bilateral. 4 crisis con clínica. Carga Fenitoína 15 mg/k. Midazolam bic 0,1. Piridoxina 100 mg/d. Midazolam 0,2 mg/k. Fenobarbital + mdz.")
5
Exámenes iniciales Examen 23/3 01,30 19,50 Na /k/cl 134/4/105
132/3,6/102 Gases PH 7,4, po2 75, pco2 31, bic 20 be -6, sat 99 PH 7,35, pco2 41, po2 69, Bic 22 be -2 Creatinina 1 0,4 Glicemia 122 Amonio 1,29 ug/ml 0,8 Láctico 12 Ca/p/mg 9/4,9/- 8,3/ Nitrógeno ureico 19 Hemograma Recuento blancos 8850, hto 45, plaquetas Leucocitos 13800, hto 48, plaquetas Pcr TP /TTPK 55/41 63/39

6
Informe eeg EEG 24/3/14: (1) severamente alterado,
trazado base desorganizado, actividad epileptiforme centro temporal bilateral independiente. Registro de paroxismos generalizados de alto voltaje frecuentes periodos de depresión de voltaje post descarga. Se registran 4 crisis de inicio generalizada que luego focalizan zonas centro temporal derecha (2) e izquierda (2), con correlato clínico
 e izquierda (2), con correlato clínico.")
7
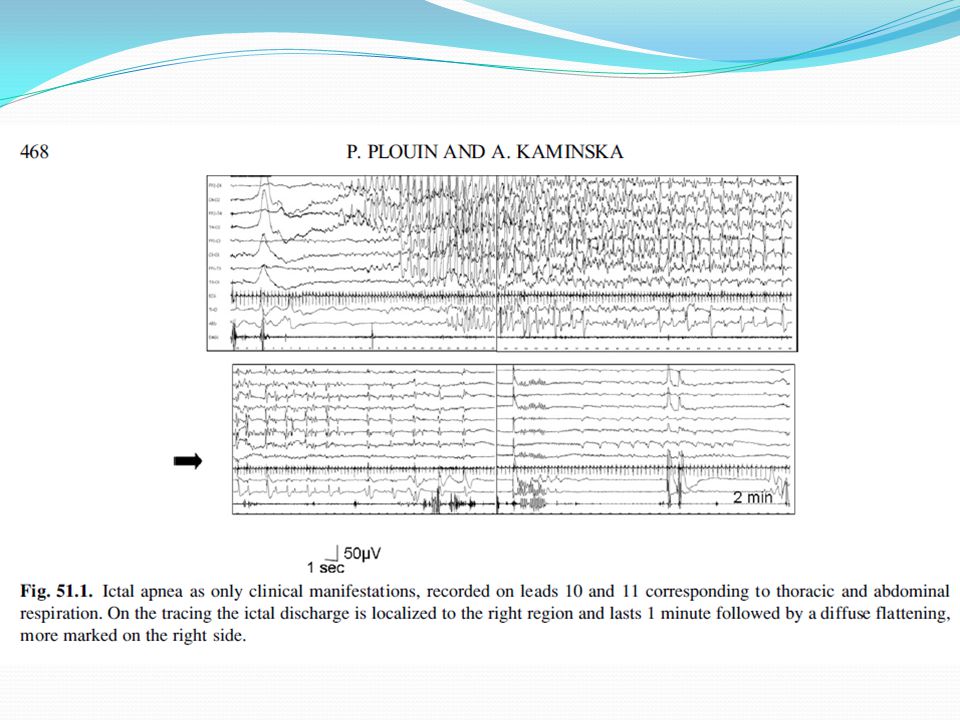
Foto eeg

8
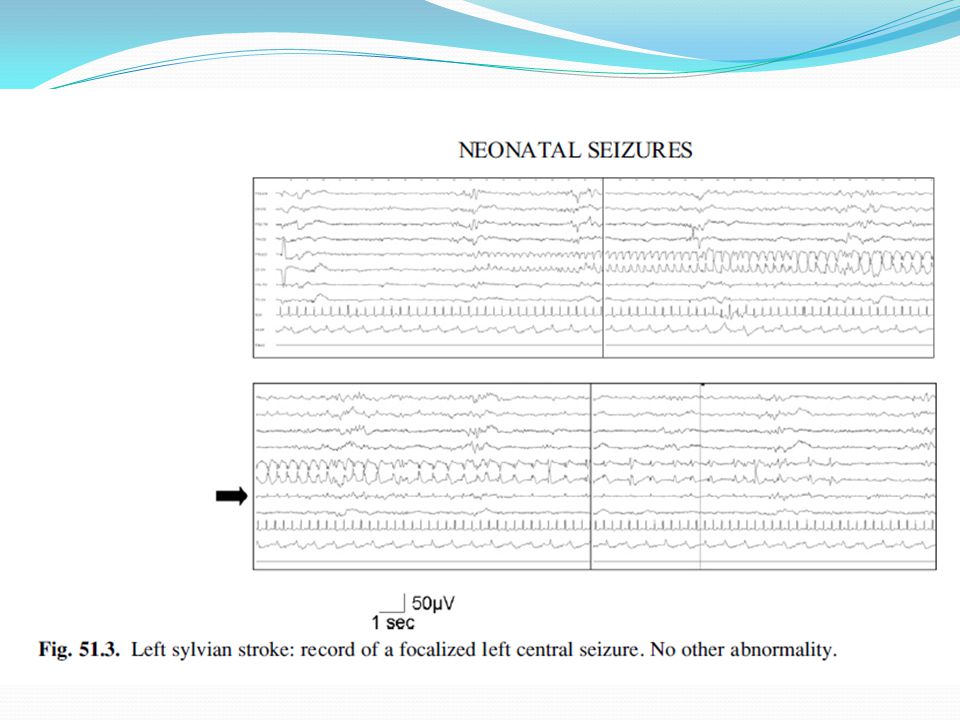
Foto eeg

10
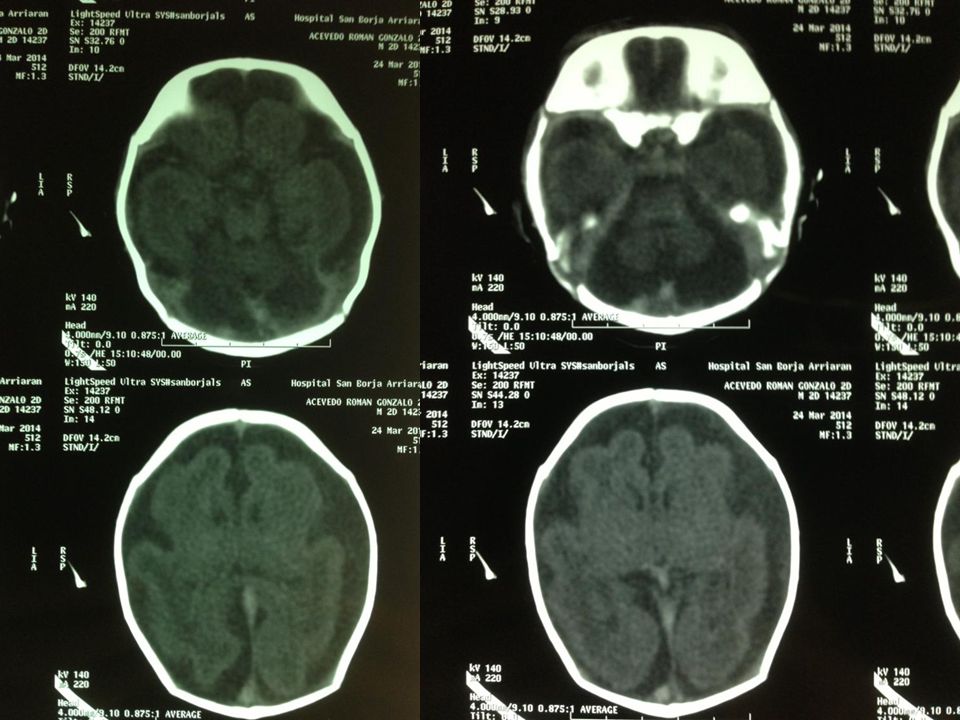
Tac cerebral 24/3

11
ATB x neumonía asociada a ventilación mecánica Aminoacidemia
G.J.A RNT PEG 22/3/2014 – 329 Apgar: 7-8 Sin crisis Crisis LZP sos Crisis 3-4 /día pero mas cortas 7-8 días 9-12 días 2-6 días 2 horas UCI NEO: en VM En VM En VM Evaluación multi sistema Cardio: HTP leve, refluj Tricuspideo Oftalmo: fondo ojo N Infecto: sugiere TORCH Genética: cariograma Endocr: sin patología TORCH negativo Niveles FB y FNT normal ATB x neumonía asociada a ventilación mecánica Aminoacidemia Aminoaciduria Láctico Amonio todo normal LZP-FB-piridoxina FNT- levetiracetam MDZ-FB-FNT-piridoxina MDZ $ ; LZP sos FB-FNT-piridoxina

12
Status: 4 crisis > 5 min
G.J.A RNT AEG 22/3/2014 – 329 Apgar: 7-8 Crisis 2-3 día Sin crisis Status: 4 crisis > 5 min 13-15 días 2 horas 16-21 días 22-26 días UCI NEO: en VM Extubación: cpap Reconexión a VM Eeg control (3) Cambio atb a cipro X cultivo + serratia m Pendiente cariograma y chagas aun tazonan Eeg control 4 LZP sos--piridoxina LVT- mdz bic 0,3 $ FB Primidona- lidocaína FB-LVT-piridoxina LZP sos LZP-FB-LVT-piridoxina $ Fenitoína
 Cambio atb a cipro. X cultivo + serratia m. Pendiente cariograma y chagas aun. tazonan. Eeg control 4. LZP sos--piridoxina. LVT- mdz bic 0,3. $ FB. Primidona- lidocaína. FB-LVT-piridoxina. LZP sos. LZP-FB-LVT-piridoxina. $ Fenitoína.")
13
Informe eeg EEG 14/4/14: (4) Severamente anormal, crisis eléctricas subintrantes de inicio focal central izquierdo (C3) con generalización secundaria y generalizadas con máxima negatividad central Frecuentes paroxismos generalizados con máxima negatividad central con periodos electrodecrementales post carga Actividad epileptiforme multifocal independiente foco predominante central izquierdo
 con generalización secundaria y generalizadas con máxima negatividad central. Frecuentes paroxismos generalizados con máxima negatividad central con periodos electrodecrementales post carga. Actividad epileptiforme multifocal independiente foco predominante central izquierdo.")
14
Foto eeg (4)
")
15
Foto eeg (4)
")
16
Diagnósticos: Sindromatico Localizatorio Etiológico Generales
Síndrome convulsivo neonatal Status epiléptico neonatal Localizatorio Cerebral difuso Etiológico Microlisencefalia Hipoplasia cerebelosa Generales RNT PEG – exposición a tóxicos antenatales Insuficiencia respiratoria Síndrome apneico Neumonía asociada a ventilación mecánica

17
Introducción Las convulsiones neonatales (CNN) son el evento neurológico mas frecuente en el recién nacido Puede ser la manifestación clínica de eventos prenatales, peri parto o post natal del SNC Son un signo marcador de disfunción de SNC que requiere diagnostico y tratamiento urgente Neurología del recién nacido, 5ª edición, Volpe: Capitulo 5, Convulsiones neonatales,

18
Definición Descarga paroxística repetitiva y estereotipada de un grupo de neuronas dentro de los primeros 28 días de vida de un RNT o hasta 44 semanas corregidas en RNPT Duración al menos 10 segundos, patrón repetido –evolutivo con amplitud mínima de 2 uV (peak a peak) Uno o mas canales del EEG Altera función cerebral EEG Gold standard Eventos que duran < 10 segundos: descarga rítmica corta (BRD) o intermitentes (BIRD) neurodesarrollo ? Neurología del recién nacido, 5ª edición, Volpe: Capitulo 5, Convulsiones neonatales,
 Uno o mas canales del EEG. Altera función cerebral. EEG Gold standard. Eventos que duran < 10 segundos: descarga rítmica corta (BRD) o intermitentes (BIRD) neurodesarrollo Neurología del recién nacido, 5ª edición, Volpe: Capitulo 5, Convulsiones neonatales,")
20
CNN: Generalidades Las convulsiones neonatales se caracterizan por tener patrones muy poco organizados y difíciles de reconocer Fundamental monitoreo en EEG Representan una emergencia medica Puede ser manifestación de enfermedad grave/tratamiento especifico Produce daño per se, por disminución de reserva energética cerebral y liberación de neurotransmisores excitatorios Episodio convulsivo puede desestabilizar situación clínica del recién nacido Neurología del recién nacido, 5ª edición, Volpe: Capitulo 5, Convulsiones neonatales,

21
CNN: Generalidades La mayor parte de las convulsiones se originan de regiones central- temporal o vertex Recomendación ACNS para eeg: p3-p4 (canal único) y c3-p3 y c4-p4 (doble canal eeg) Foco frontal y occipital son raros sugiere lesión focal Evolución en frecuencia, morfología, ubicación ayuda a diferenciar convulsiones de patrones rítmicos no ictales y descargas Neurología del recién nacido, 5ª edición, Volpe: Capitulo 5, Convulsiones neonatales,
 y c3-p3 y c4-p4 (doble canal eeg) Foco frontal y occipital son raros sugiere lesión focal. Evolución en frecuencia, morfología, ubicación ayuda a diferenciar convulsiones de patrones rítmicos no ictales y descargas. Neurología del recién nacido, 5ª edición, Volpe: Capitulo 5, Convulsiones neonatales,")
22
Incidencia Estudios poblacionales sugieren incidencia de 1-3/1000 de los recién nacidos vivos Sin embargo aumenta en población seleccionadas 11-30% en prematuros (inversamente proporcional a EG) La mayor parte ocurre durante la primera semana (80%) Saliba et al 1999; Silverstein y Jensen, 2007
 La mayor parte ocurre durante la primera semana (80%) Saliba et al 1999; Silverstein y Jensen,")
23
Fisiopatología Crisis descarga eléctrica repentina, sincrónica y excesiva de un grupo neuronal Despolarización causada por Disminución de producción energía y falla bomba Na-K Liberación excesiva de glutamato y consumo reducido células Déficit de neurotransmisor inhibitorios: Gaba, déficit piridoxina Hipocalcemia e hipomagnesemia: inhibe el traspaso de Na a las neuronas Neurología del recién nacido, 5ª edición, Volpe: Capitulo 5, Convulsiones neonatales,

24
Fisiopatología Crisis RN son diferentes a otras etapas de la vida
El desarrollo de estructuras subcorticales y límbicas están relativamente avanzadas La mielinización y fonación sinapsis corteza cerebral están relativamente incompletos mas frecuente crisis sutiles, apneas

25
Fisiopatología Receptor glutamato: sinapsis, sitios no sinápticos, neuronas y células gliales 3 tipos inotrópicos: NMDA, AMPA, kainato En RN NMDA y AMPA están sobre expresados, y además su composición lo hace mas excitables Antagonista NMDA Ketamina, meperidina (pero sedantes) Antagonista AMPA topiramato
 Antagonista AMPA. topiramato.")
26
Fisiopatología Receptor GABA: “compensa” excitabilidad, pero hay poco en EG precoz Además su activación produce excitación en vez de inhibición (por homeostasis canales cloro) desbalance entre NKCC1 y KCC2 Maduración de caudal a rostral (manifestación motora y luego eléctrica) Neurología del recién nacido, 5ª edición, Volpe: Capitulo 5, Convulsiones neonatales,
 desbalance entre NKCC1 y KCC2. Maduración de caudal a rostral (manifestación motora y luego eléctrica) Neurología del recién nacido, 5ª edición, Volpe: Capitulo 5, Convulsiones neonatales,")
27
Jensen, update on mechanism and management cl perinatology, 2009 881-900

28
Clasificación Ocasionales: Epilepsias verdaderas
Cuadro agudo por agresión puntual SNC (90%) EJ: encefalopatía hipóxica isquémica, trastorno cerebro vascular, infecciones SNC, trastornos metabólicos transitorios 10-20% evolucionaran a epilepsia Epilepsias verdaderas Crisis recidivantes con expresión EEG definida Etiología (la mayor parte) desconocida EJ: convulsión idiopática benigna, epilepsia neonatal sintomática, convulsión familiar benigna neonatal, encefalopatía mioclónica precoz Neurología del recién nacido, 5ª edición, Volpe: Capitulo 5, Convulsiones neonatales,
 EJ: encefalopatía hipóxica isquémica, trastorno cerebro vascular, infecciones SNC, trastornos metabólicos transitorios % evolucionaran a epilepsia. Epilepsias verdaderas. Crisis recidivantes con expresión EEG definida. Etiología (la mayor parte) desconocida. EJ: convulsión idiopática benigna, epilepsia neonatal sintomática, convulsión familiar benigna neonatal, encefalopatía mioclónica precoz. Neurología del recién nacido, 5ª edición, Volpe: Capitulo 5, Convulsiones neonatales,")
29
Clasificación (VOLPE)
clónicas tónicas Tipo convulsión mioclónica sutiles

30
Clasificación: Clónicas
La crisis típica del RNT. (25-30%) Movimientos rítmicos y lentos con correlato eeg Pueden ser focales, repetidas y localizadas Habitualmente no hay compromiso conciencia No implican daño local Multifocal: tiene carácter migratorio, de una extremidad a otra, tiene correlato eeg y pueden ser causadas por un compromiso severo SNC
 Movimientos rítmicos y lentos con correlato eeg. Pueden ser focales, repetidas y localizadas. Habitualmente no hay compromiso conciencia. No implican daño local. Multifocal: tiene carácter migratorio, de una extremidad a otra, tiene correlato eeg y pueden ser causadas por un compromiso severo SNC.")
31
Clasificación: Tónicas
5 % de las CNN Movimientos lentos y sostenidos Tiene correlato eeg Pronostico reservado Puede ser focal (cambio mantenido de 1 extremidad o tronco, apnea o cianosis) generalizada (mov brusco de flexión de eess y extensión/abducción –aducción eeii) o flexo extensión 4 extremidades simula descerebración/decorticación
 generalizada (mov brusco de flexión de eess y extensión/abducción –aducción eeii) o flexo extensión 4 extremidades. simula descerebración/decorticación.")
32
Clasificación: Mioclónica
15-20% de las CNN Movimientos flexión brusca o sacudidas de extremidades Focal o generalizadas Generalmente secundaria a patología difusa cerebro Importante descartar patología metabólica

33
Clasificación: Sutiles
50% de las CNN Pueden presentarse junto a las otras No tienen correlato eeg Pueden no ceder con tratamiento anticonvulsivo EJ: parpadeo, ojos permanentemente abiertos, chupeteo, pedaleo, natatorios, fenómenos autonómicos (hipertensión, taquicardia, bradicardia), apnea
, apnea.")
35
Diagnostico diferencial
Temblores Sin fenómenos oculares, orobucales, autonómicos Se suprime al sujetar extremidad Ojo con hipoglicemia, hipocalcemia, depravación, EHI Mioclonías benignas del sueño Movimientos mioclónicos que solo ocurren durante sueño. Pueden durar varios minutos. EEG normal Se resuelve después de los 2 meses Hiperekplexia Respuesta sobresalto exagerada a la manipulación y estimulo auditivo/visual Autosómico dominante hiperexcitabilidad neuronas reticulares Desaparece después de los 2 años Neurología del recién nacido, 5ª edición, Volpe: Capitulo 5, Convulsiones neonatales,

36
Etiología Encefalopatía hipóxico isquémica: (50% CNN)
En general 1as 24 horas de vida Trastorno cerebro vasculares (12-20% de las CNN) Hemorragia intracraneana: Intraventricular: > RNPT; en primeros 3 días de vida. Subaracnoideas / Subdurales: >en RNT Lesión isquémica cerebral: es la 2ª causa CNN Trombosis in situ /embolismo placenta –cardiaco Amerita estudio hipecoagulabilidad (prot c-s- factor v leiden)
 Hemorragia intracraneana: Intraventricular: > RNPT; en primeros 3 días de vida. Subaracnoideas / Subdurales: >en RNT. Lesión isquémica cerebral: es la 2ª causa CNN. Trombosis in situ /embolismo placenta –cardiaco. Amerita estudio hipecoagulabilidad (prot c-s- factor v leiden)")
37
Crisis sub clinica izquierda RN asfixiado grave

40
Etiología Alteraciones metabólicas transitorias (5-20%)
Hipoglicemia, hipocalcemia, hipernatremia, hiponatremia, hipomagnesemia Malformaciones SNC (5-10%) Infecciones SNC (3-20%) Sospecha en RN febril o con factores de riesgo para infección SNC PL mandatoria para diagnóstico/tratamiento
 Infecciones SNC (3-20%) Sospecha en RN febril o con factores de riesgo para infección SNC. PL mandatoria para diagnóstico/tratamiento.")
41
Etiología Errores congénitos del metabolismo
periodo libre de síntomas intoxicación Rechazo alimentario, letargia, trastorno respiratorio, convulsiones Los mas frecuentes son alteración metabolismo aminoácidos/ciclo de urea (amonio elevado, anión gap elevado, cetonemia, Hiperglicinemia no cetosica solo convulsiones Síndrome depravación drogas Desconocido: 10%

42
Neurología del recién nacido, 5ª edición, Volpe: Capitulo 5, Convulsiones neonatales,

43
Factores de riesgo para CNN
Maternos Intraparto Recién nacido Edad materna > 40 años Sufrimiento fetal agudo RNPT Nulípara Desprendimiento placenta, prolapso cordón RN bajo peso DM gestacional o pre gestacional Fiebre materna, corioamniolitis RN post termino Sexo masculino Vasudevan, Semanas in fetal and neonatal medicina 18 (2013)
")
44
Síndromes epilépticos neonatales
Existen pocos identificados en este periodo A la fecha son 4 (2 benignos y 2 malignos): Convulsión idiopática neonatal convulsión benigna familiar encefalopatía epiléptica neonatal con estallido supresión: encefalopatía mioclónica precoz encefalopatía epiléptica infantil precoz o síndrome Ohtahara
: Convulsión idiopática neonatal. convulsión benigna familiar. encefalopatía epiléptica neonatal con estallido supresión: encefalopatía mioclónica precoz. encefalopatía epiléptica infantil precoz o síndrome Ohtahara.")
45
Síndromes epilépticos neonatales
Convulsiones benignas familiar Herencia dominante Diagnostico exclusión RNT – peso normal –Apgar >7 Luego de intervalo libre primeras convulsiones inician en día 2 o 3 de vida. Pueden durar 1-6 meses Condición neurológica normal. (a lo mas hipotonía transitoria) Crisis son aisladas, duran 1-3 minutos, pueden recurrir Se han identificado genes que codifican canal potasio KCNQ2 (cromosoma 20q) + KCNQ3 (cromosoma 8q)
 Crisis son aisladas, duran 1-3 minutos, pueden recurrir. Se han identificado genes que codifican canal potasio KCNQ2 (cromosoma 20q) + KCNQ3 (cromosoma 8q)")
46
Síndromes epilépticos neonatales
Convulsiones neonatales idiopática benignas: Reportado en Australia y Francia 1977 Prevalencia 4-38% en otros reportes. probable solo 4% Primeras convulsiones entre día 1-7 (y 90% entre 4-6) Crisis multifocales clónicas, parciales, alterna lados, apneas, nunca tónicas. Rara vez generalizadas. Duran 1-3 minutos. EEG interictal es normal, discontinuo o alterado por alteraciones focales o multifocales. 60% presenta alternancia theta punta Relacionado con niveles bajos de Zinc
 Crisis multifocales clónicas, parciales, alterna lados, apneas, nunca tónicas. Rara vez generalizadas. Duran 1-3 minutos. EEG interictal es normal, discontinuo o alterado por alteraciones focales o multifocales. 60% presenta alternancia theta punta. Relacionado con niveles bajos de Zinc.")
47
Síndromes epilépticos neonatales
Encefalopatía mioclónica precoz Inicio 1ª semana Causa desconocida EEG patrón estallido supresión Persiste mas allá del 1er años de vida Mal pronostico Encefalopatía epiléptica infantil precoz (sd. Ohtahara) Inicio primeros 3 meses Acompañados de encefalopatía severa Resistente a tratamiento Mal pronostico (muere o RDSM severo)
 Inicio primeros 3 meses. Acompañados de encefalopatía severa. Resistente a tratamiento. Mal pronostico (muere o RDSM severo)")
48
Diagnostico Anamnesis: antecedentes maternos, familiares,
embarazo y parto, monitorización en parto, tipo de parto, características LA, Apgar, gases cordón

49
Diagnostico Examen físico completo: Descripción del evento
Existen estudios concordancia neo/staff/neuro/eeg no buena correlación (Malone 2009) 50% 80-90% de convulsiones neonatales al EEG no tienen correlato clínico (Bye y flanagan 1995; Clancy ; Lawrence 2009, otros) Convulsiones focales clónicas tienen mejor pronostico en gral Convulsiones sutiles/subclínicas peor pronostico
 50% 80-90% de convulsiones neonatales al EEG no tienen correlato clínico (Bye y flanagan 1995; Clancy ; Lawrence 2009, otros) Convulsiones focales clónicas tienen mejor pronostico en gral. Convulsiones sutiles/subclínicas peor pronostico.")
50
Diagnostico Laboratorio básico:
Sangre: glicemia, calcemia, mg, elp, gases, láctico, amonio, hemograma, pcr, hemocultivos, tándem mass Orina: aminoaciduria, Screening metabólico orina LCR: cultivos, cito químico, amonio, acido láctico, estudio metabólico

52
Diagnostico Neuroimagen EEG: Ecografía cerebral Tac cerebral RNM
Monitorización es primordial (disociación electro clínica) EEG 12 canales Gold estándar aEEG continuo, útil en RNT y RNPT Según zona de crisis, puede no detectar. No reemplaza EEG
 EEG 12 canales Gold estándar. aEEG continuo, útil en RNT y RNPT. Según zona de crisis, puede no detectar. No reemplaza EEG.")
53
EEG en RN Interpretación es diferente que en niños mas grandes/adultos
Ausencia de patrones típicos de convulsión no descarta crisis origen cortical Siempre realizar con piridoxina Características Interictales: presencia de onda punta focales intermitente temporales pueden ser normales Ritmo de base: informa sobre grado severidad lesion EEG característicos

54
¿Qué niños deben ser monitorizados?
RN con clínica sugerente convulsiones RN con antecedentes de evento hipóxico- isquémica RN con EHI moderada severa Rn en hipotermia (especialmente en recalentamiento) RN con Neuroimagen alterada RNPT de extremo bajo peso (<1000)
 RN con Neuroimagen alterada. RNPT de extremo bajo peso (<1000)")
58
Tratamiento: Generalidades
El cerebro del RN es mas propenso a las crisis Crisis prolongadas y recurrentes perjudican el cerebro en desarrollo Retraso de perdida neuronal Disminución neurogenesis Reorganización sináptica Cambios en la plasticidad del hipocampo Disminución densidad dendritas en neuronas Patel, 2011: recognition and management of neonatal seizurez, paediatrics and Child health

59
Tratamiento General: Especifica anticonvulsiva:
vía venosa, estabilización funciones vitales, metabólicas, iniciar terapia especifica si corresponde (SG, ATB, etc..) No requiere eeg para iniciarse Especifica anticonvulsiva: No hay estudios con suficiente # pacientes sobre una superioridad absoluta de 1 droga sobre otra La terapia + usada fenobarbital Balancear beneficios vs efectos adversos/riesgos
 No requiere eeg para iniciarse. Especifica anticonvulsiva: No hay estudios con suficiente # pacientes sobre una superioridad absoluta de 1 droga sobre otra. La terapia + usada fenobarbital. Balancear beneficios vs efectos adversos/riesgos.")
61
Tratamiento – drogas AC
Fenobarbital sódico: (primera línea) Agonista GABA carga 20 mg/k/dosis en 10 minutos. Si persisten convulsiones repetir a los 15 minutos Mantención 3-5 mg/k/d, fraccionado cada 12 o 24 horas Controla con éxito 50% crisis eléctricas Larga vida media (2-4 días), penetra BHE Mantención en RNPT < 28 semanas mas errática, requiere niveles
 Agonista GABA. carga 20 mg/k/dosis en 10 minutos. Si persisten convulsiones repetir a los 15 minutos. Mantención 3-5 mg/k/d, fraccionado cada 12 o 24 horas. Controla con éxito 50% crisis eléctricas. Larga vida media (2-4 días), penetra BHE. Mantención en RNPT < 28 semanas mas errática, requiere niveles.")
62
Tratamiento – drogas AC
Fenitoína: Reduce conductancia eléctrica neuronal mediante estabilización canales de sodio Carga 20 mg/k/dosis, lento minutos (depresión cardiaca) Dosis mantención 5 mg/k/d (ev.) oral no por errática Combinación FB + FNT control convulsión 65-70%
 Dosis mantención 5 mg/k/d (ev.) oral no por errática. Combinación FB + FNT control convulsión 65-70%")
63
Tratamiento – drogas AC
Levetiracetam Cada vez mas usado pro su efectividad. No se han descrito efectos adversos Mecanismo desconocido No requiere medición niveles plasmáticos Puede ser usado como monoterapia mantención Carga 50 mg/k, mantención mg/k/día fraccionado c/12 horas

64
Tratamiento – drogas AC
lidocaína Bloquea la entrada de Na a la neurona Opción de segunda línea (Suecia). Yugula convulsiones 70-92% Dosis 2 mg/k carga 30 minutos, luego 6 mg/k/h por 12 horas. Disminuir dosis en 2 mg/k/h según respuesta c/12 horas hasta suspender Efectos adversos: arritmias (4-8% puede tener arritmia durante BIC), hipotensión No indicarla si se ha recibido Fenitoína
. Yugula convulsiones 70-92% Dosis 2 mg/k carga 30 minutos, luego 6 mg/k/h por 12 horas. Disminuir dosis en 2 mg/k/h según respuesta c/12 horas hasta suspender. Efectos adversos: arritmias (4-8% puede tener arritmia durante BIC), hipotensión. No indicarla si se ha recibido Fenitoína.")
66
Tratamiento – drogas AC
benzodiacepinas Agonistas GABA Clonazepam: dosis 0,01 mg/k (vida media horas) Midazolam: bolo 0,1 -0,2 mg/k y luego infusión continua 0,05-0,4 mg/k/hora. Si responde mantener 24 horas y disminuir lentamente Puede producir Mioclonías Vida media 6 horas Lorazepam: dosis 0,05-0,1 mg/k. efecto inicio a los 2-3 min Duración efecto 6-24 horas Piridoxina: 100 mg por 3 días. Si hay respuesta mantener
 Midazolam: bolo 0,1 -0,2 mg/k y luego infusión continua 0,05-0,4 mg/k/hora. Si responde mantener 24 horas y disminuir lentamente. Puede producir Mioclonías. Vida media 6 horas. Lorazepam: dosis 0,05-0,1 mg/k. efecto inicio a los 2-3 min. Duración efecto 6-24 horas. Piridoxina: 100 mg por 3 días. Si hay respuesta mantener.")
70
Otras terapias Topiramato Bumetanida
Antagonista glutamato mediante bloqueo receptor AMPA y bloqueador canal Na Se ha utilizado con éxito en RNT en hipotermia Bumetanida Diurético asa 40x mas potente que furosemida. Disminuye la concentración intracelular de cloro disminuye acción depolarizante de GABA

71
Otras terapias Acido valproico carbamazepina Piridoxina:
En convulsiones intratables Riesgo hiperamonemia/hepatotoxicidad limitan en RN carbamazepina Eficaz en control convulsiones Absorción oral es muy errática en RNPT Piridoxina: Sospecha en crisis clónicas multifocales resistentes tratamiento mg ev bajo monitorización EEG Si hay respuesta mantener Acido folínico: Leucovorina 2,5 mg/ev Si no responde a piridoxina

72
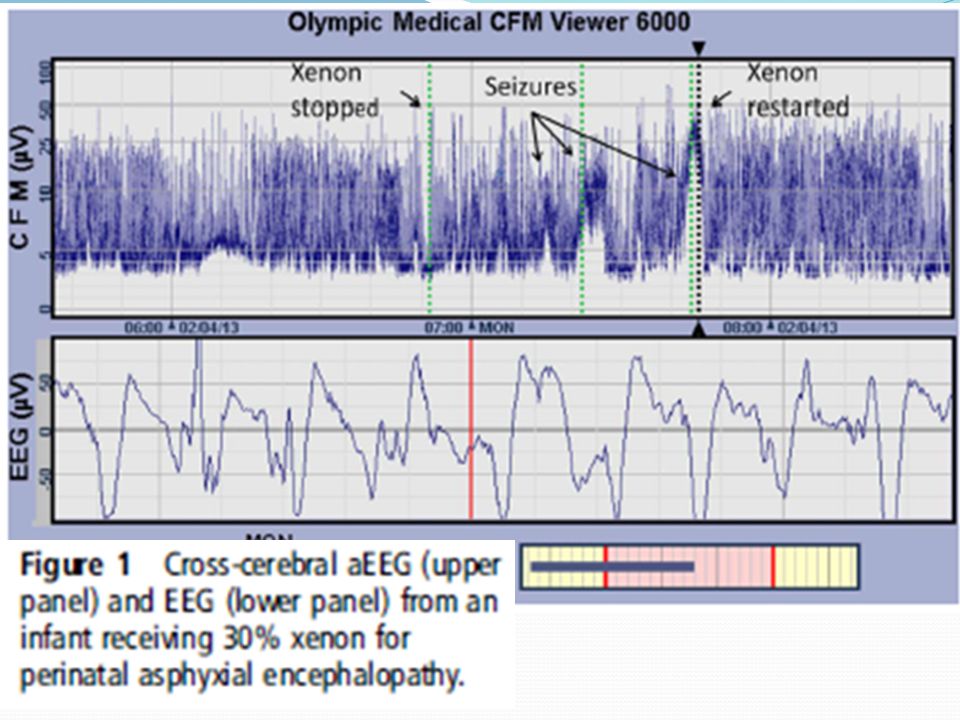
tratamiento Xenón? Gas monoatómico, altamente soluble
Inhibición no competitiva de NMDA, efecto anti apoptótico y neuroprotector Estudio en RNT asfixiados con EHI en hipotermia + aEEG: convulsiones cesan durante la administración, reaparecen tras suspensión

74
Duración tratamiento No hay consenso sobre duración una vez que crisis han cedido Medicamentos AC tienen efectos adversos, pero además en animales favorecen apoptosis neuronal Riesgo recurrencia: <10% lactantes con EEG normal > 50% con examen neurológico anormal y/o EEG con actividad de base anormal

75
Pronostico Mortalidad CNN ha disminuido
Trastorno en neurodesarrollo 10-20% de los niños con CNN

76
Status Epiléptico Neonatal

77
SEN Emergencia neurológica
Definido en pediatría/medicina como crisis epiléptica que dure mas de 30 minutos o varias crisis sin que haya recuperación completa de conciencia En neo no tan clara definición La mayoría de las crisis en RN duran < 5 min Por clínica no puedo asegurar que entre crisis el RNPT vuelva a “normal” (crisis sutiles, etc.) El RNPT si bien al parecer resiste convulsiones mejor, el efecto multi sistémico no siempre es así
 El RNPT si bien al parecer resiste convulsiones mejor, el efecto multi sistémico no siempre es así.")
78
SEN Otras definiciones: mas del 50% de un determinado tiempo de observación, 5 minutos, 10 minutos. En HCSBA: usa definición operativa de crisis única o varias de mas de 5 min de duración sin recuperación entre ellas Incidencia: 20/100,000 habitantes Distribución bimodal, tasas mas altas el 1er año y >60 años En neo desconocida subdiagnosticado? aEEG, video EEG

79
SEN Causas: febril, idiopática, infecciosa, errores innatos metabolismo, tóxicos, abuso sustancias, tumores cerebrales, trauma, enfermedades cerebro vascular, hipoxia/anoxia Además en pacientes con pacientes epilepsia y tratamientos crónicos que suspenden bruscamente terapia. Sin causa 10-30%

80
SEN tratamiento: febril, idiopática, infecciosa, errores innatos metabolismo, tóxicos, abuso sustancias, tumores cerebrales, trauma, enfermedades cerebro vascular, hipoxia/anoxia Además en pacientes con pacientes epilepsia y tratamientos crónicos que suspenden bruscamente terapia. Sin causa 10-30%

81
SEN Tratamiento: 3°

82
SEN 1°

83
Microcefalia y Lisencefalia

84
Introducción Las malformaciones cerebrales son una causa significativa de morbilidad neurológica en niños Abarcan numerosos trastornos difícil clasificarlos. Clasificación actual basada en etapa de desarrollo cerebral Trastorno proliferación celular (microcefalia) Trastorno migración celular (Lisencefalia) Trastorno organización cortical
 Trastorno migración celular (Lisencefalia) Trastorno organización cortical.")
85
Microcefalia Descrita por Giacomini en 1885
Condición en la que el cerebro no logra un crecimiento normal (mas una observación clínica que diagnostico) Definición: Microcefalia menor a – 3 DE (ajustado por edad/sexo) Discapacidad intelectual grado variable Ausencia de otro trastorno neurológico o de crecimiento
 Definición: Microcefalia menor a – 3 DE (ajustado por edad/sexo) Discapacidad intelectual grado variable. Ausencia de otro trastorno neurológico o de crecimiento.")
86
Microcefalia Clásicamente se presenta al momento de nacimiento
Puede llegar a ser cc -12 DE Epilepsia es poco común (pero se ha reportado y no es criterio de exclusión) Tiene patrón herencia autosómica recesivo, en la ultimas décadas se ha comprendido mas heterogenicidad genética
 Tiene patrón herencia autosómica recesivo, en la ultimas décadas se ha comprendido mas heterogenicidad genética.")
87
Microcefalia Se han descrito 8 (al menos) loci con esta condición llamados MCPH1 (o microcefalina) hasta 8 Y se han descrito 5 genes asociados: MCPH1, CDK5RAP2, ASPM, CENPJ Y STILL No hay clara correlación genotipo-fenotipo, por lo que la clínica de un especifico caso no es posible saber cual gen esta predominando Radiológicamente la estructura cerebral esta generalmente conservada, pero con simplificación del patrón de giro en pacientes con ASPM y MCPH1

88
Microcefalia Se han descrito además heterotopias nodulares peri ventriculares en familias con MCPH1, aunque no es exclusivo para MCPH1 ASPM (MCPH5) es la causa mas común de microcefalia. Reportado en 40% de casos con origen pakistaní, árabe o europeo Mas de 50 mutaciones ASPM descritas Existen otras mutaciones independientes descritas para otros genes de microcefalia, pero su espectro clínico esta menos definido
 es la causa mas común de microcefalia. Reportado en 40% de casos con origen pakistaní, árabe o europeo. Mas de 50 mutaciones ASPM descritas. Existen otras mutaciones independientes descritas para otros genes de microcefalia, pero su espectro clínico esta menos definido.")
92
Microcefalia Los genes descritos en microcefalia participan en división celular y regulación de ciclo celular ASPM, CDK5RAP2, CENPJ Y STILL codifican proteínas centrosoma ASPM homologo para gen Drosophila asp (huso anormal): proteína mantención integridad centrosoma y huso mitótico (modelo animal) Además es importante en la mantención de la orientación en el plano de división celular en progenitores neurales depleción precoz de células progenitoras insuf neurona (microcefalia)
: proteína mantención integridad centrosoma y huso mitótico (modelo animal) Además es importante en la mantención de la orientación en el plano de división celular en progenitores neurales depleción precoz de células progenitoras insuf neurona (microcefalia)")
93
Microcefalia CDK5RAP2: también participa en mantención de cohesión de centrosoma MCPH1 esta implicada en la reparación de ADN y condensación de cromosomas Parcialmente tiene la misma vía de señal que ATR (ataxia-telangectasia-RAD3 relacionado), el cual esta mutado en algunos Síndrome Sequel (microcefalia, talla baja y dismorfias faciales) Función en localización y división de centrosoma ¿rol en factores de crecimiento/división cerebral?
, el cual esta mutado en algunos Síndrome Sequel (microcefalia, talla baja y dismorfias faciales) Función en localización y división de centrosoma. ¿rol en factores de crecimiento/división cerebral")
94
Lisencefalia Grupo de trastornos caracterizados por una corteza anormalmente liso Dividido en 2 categorías: clásica (tipo 1), y empedrado (tipo 2) Empedrado: defecto bioquímico O-glicosilacion de α-dextroglicano Compromete integridad de superficie pial sobre migración de neuronas corticales a través de superficie pial Estas heterotopias forman nódulos “empedrados” en la superficie cerebral Otros “empedrados” en Walker-Warburg, síndrome musculo-ojo, distrofia muscular Fukuyama

95
Lisencefalia En Lisencefalia clásica, la integridad de la superficie pial esta intacta, pero la corteza cerebral esta anormalmente engrosada Y la estructura de 6 capas severamente comprometida El primer gen identificado fue LIS1: localizado en cromosoma 17p13.3. dos entidades clínicas asociadas: síndrome Miller Dieker y secuencia Lisencefalia aislada

96
SMD: dismorfia facial (frente prominente, hueco bitemporal, nariz corta con narinas hacia arriba, labio superior prominente, comisuras delgadas, maxilar pequeño) ILS: solo Lisencefalia, sin características de SMD

97
Lisencefalia ILS deleción pequeña o mutación LIS1.
MDS + grande: involucra LIS1 y 40 genes vecinos La mayor parte son mutaciones de novo (baja recurrencia) Pero se han descrito casos con translocaciones balanceadas (familias) DCX (doblecortina): gen ubicado en cromosoma X Causa Lisencefalia en hombres (hemicigotos), en mujeres casos mas leves (bandas heterotopias subcorticales) LIS1 y DCX ¾ de los casos de ILS
 Pero se han descrito casos con translocaciones balanceadas (familias) DCX (doblecortina): gen ubicado en cromosoma X. Causa Lisencefalia en hombres (hemicigotos), en mujeres casos mas leves (bandas heterotopias subcorticales) LIS1 y DCX ¾ de los casos de ILS.")
98
Lisencefalia TUBA1A: gen que se encuentra mutado en pacientes con Lisencefalia clásica (4% casos) Microcefalia congénita, diplejía espástica o cuadriplejia, discapacidad intelectual. Existen casos mas leves compromiso lenguaje, logran marcha Son mutaciones habitualmente de novo, pero su patrón de herencia es de autosómico domínate Otros genes: RELN, VLDLR y ARX: “variantes de Lisencefalia”: no comparten características rx con LIS1, DCX, TUBA1A

99
Lisencefalia Funciones gen Lisencefalia: relacionado estrechamente con micro túbulos Proteínas LIS1 interactúa con dineina y en conjunto con NDEL regula el motor micro túbulo Proteínas DCX: une micro túbulos y estabiliza polimerización Proteínas TUBA1A: componente estructural de micro túbulos (isoforma α-tubulina)
")
100
Lisencefalia Otros: RELN: proteína secretada por células Cajal-retzius en la capa 1 de la corteza cerebral. Inhibe migración neuronal (señal stop) VLDLR: actúa como receptor (junto con receptor 2 apolipotroteina E) para proteína RELN
 para proteína RELN.")
101
Clasificación malformaciones de desarrollo cortical (2012)
Malformaciones secundaria a anormalidades prolif o apoptosis neuronal y glial IA microcefalia IB megalencefalia IC disgenesia cortical y prolif sin neoplasia ID disg cortical y prolf c/ neo Grupo I Malformaciones debido a migración neuronal anormal IIA heterotopia IIB Lisencefalia IIC heterotopia subcortical y displasia sub lobar IID empedrado Grupo II IIA polimicrogiria y esquicencefalia IIC displasias subcortical IIID microcefalia post migración Malformaciones secundarias a desarrollo post migración anormal Grupo III

102
referencias

Presentaciones similares