Descargar la presentación
La descarga está en progreso. Por favor, espere

1
ESTADOS DE HIPERCOAGULABILIDAD CESAR GARCIA CASALLAS MEDICINA INTERNA FARMACOLOGIA CLINICA

2
Problemas: Confirmar el Diagnóstico. Investigar, establecer la causa.
Terapia: Warfarina, Heparina, ASA. Corto tiempo vs Largo tiempo.

6
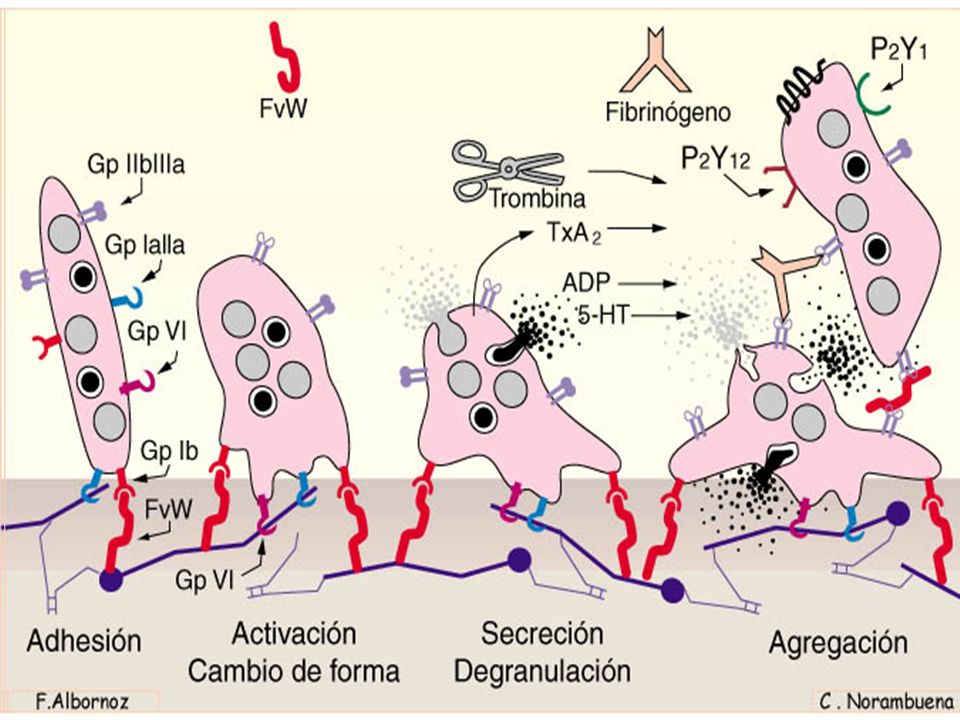
MEDIO INTERNO Hemostasia Agregación. Este mecanismo puede ponerse en marcha por varias vías metabólicas. El ADP y el TXA2 liberado por las plaquetas tiene un papel fundamental en la activación de la agregación de más plaquetas. Si la lesión es pequeña, es reversible. Endotelio vascular PGI2 Inductor de agregación Ac. Araquidónico PGG2 Tromboxano A2 TXA2 Fosoflipasa Cicloxigenasa ATP AMPc AMP Adenil ciclasa + Ca++ Membrana Plaquetar Cuerpos densos Trpmboxano sintetasa LA PLAQUETA

11
Enfermedades de la hemostasia
Síndromes hemorrágicos Hemostasia primaria Alteraciones cuantitativas de las plaquetas Trombocitopenias Alteraciones funcionales de las plaquetas Defectos extrínsecos Enfermedad de von Willebrand Defectos intrínsecos Alteraciones de agregación-secreción Hemostasia secundaria Déficit de factores de la coagulación Hemofilias Síndromes hipercoagulables Adquirida hereditaria Defecto de proteínas anticoagulantes naturales

12
Estudio básico de hemostasia para síndromes hemorrágicos
Recuento de plaquetas Tiempo de sangría Tiempo de protrombina Tiempo de tromboplastina parcial activado (TTPA)
")
14
Tiempo de Sangría

15
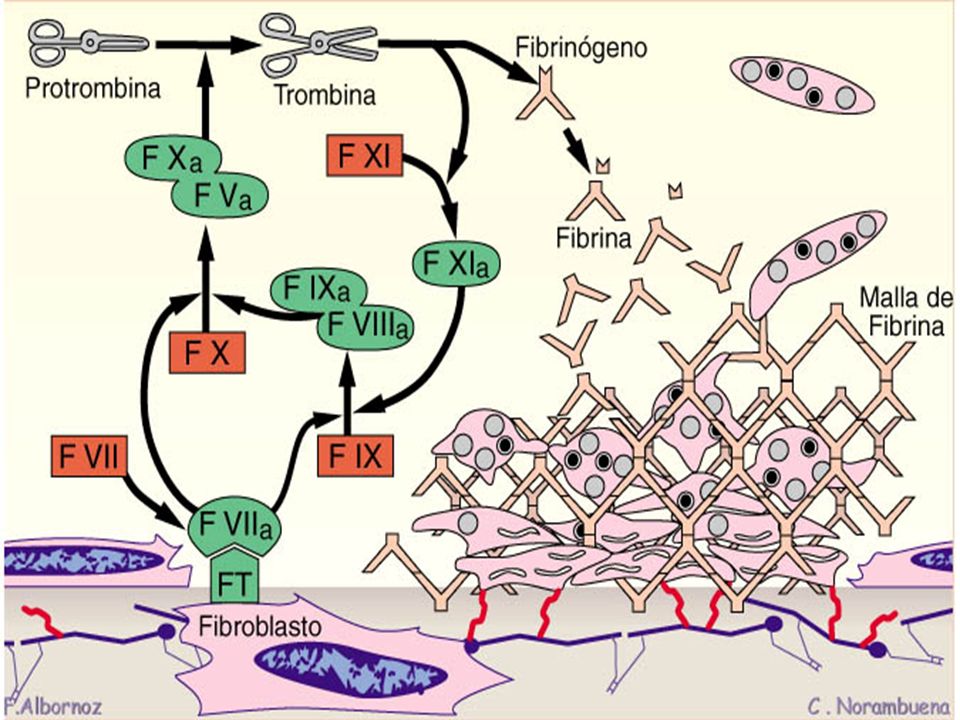
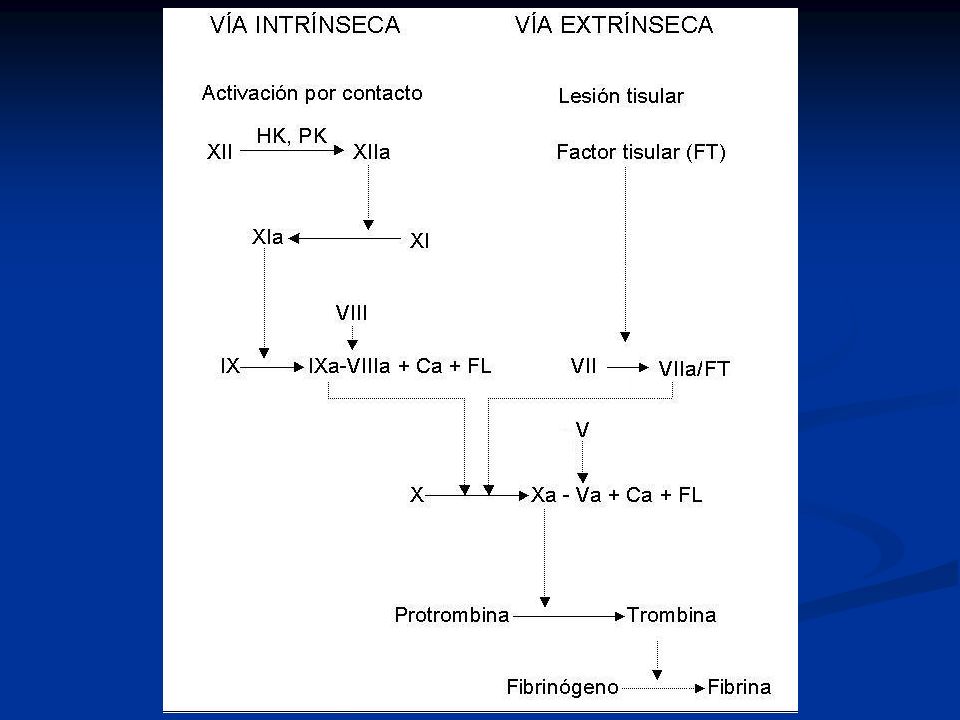
TP Fibrinógeno Trombina Protrombina Fibrina FXII FXIIa FIX FXI FXIa
FVIIa FT FL Ca++ FX FIXa FL Ca++ FVIIIa FXa FL Ca++ FVa TP Trombina Protrombina Fibrinógeno Fibrina

16
TTPA Fibrinógeno Trombina Protrombina Fibrina FXII FXIIa FIX
FXI FXIa FIX FVIIa FT FL Ca++ FX FIXa FL Ca++ FVIIIa FXa FL Ca++ FVa TTPA Trombina Protrombina Fibrinógeno Fibrina

17
Enfermedades hemorrágicas
Hemostasia primaria Alteraciones cuantitativas de las plaquetas Trombocitopenias Alteraciones funcionales de las plaquetas Defectos extrínsecos Enfermedad de von Willebrand Defectos intrínsecos Alteraciones de agregación-secreción Hemostasia secundaria Déficit de factores de la coagulación Hemofilias

18
SINDROMES HIPERCOAGULABLES

19
ESTADOS HIPERCOAGULABLES Definición
“Alteración de los mecanismos hemostáticos que predisponen a la trombosis” (BCSH). “Desarrollo de trombosis espontánea o de gravedad desproporcionada al estímulo, fenómenos tromboembólicos recurrentes o que aparecen a edad temprana (USA). Tendencia a desarrollar trombosis como consecuencia de factores predisponentes que pueden ser genéticos, adquiridos o ambos.
. Desarrollo de trombosis espontánea o de gravedad desproporcionada al estímulo, fenómenos tromboembólicos recurrentes o que aparecen a edad temprana (USA). Tendencia a desarrollar trombosis como consecuencia de factores predisponentes que pueden ser genéticos, adquiridos o ambos.")
20
Investigación: HC Historia Familiar Medicamentos (hormonas).
Evento Reciente. Examen Físico. Factores de Riesgo

21
ESTADOS HIPERCOAGULABLES
Estímulos Prothromboticos Adquiridos Mutaciones Heredadas Protromboticas Thrombosis Factor V Leiden Protrombina 20210A Proteina C deficiency Proteina S deficiency Antitrombina deficiencia Plaqueta Pagajosa Antifosfolipid Anticuerpos Malignidad Inmobilizacion Cirugía Embarazo Estrogenos Hiperhomocisteinemia Heparina-inducida Trombocitopenia

22
Factores de riesgo asociados a trombosis Arterial y/o Venosa
Venosa Arterial Diabetes Tabaquismo Hipertensión Hipercolesterolemia Hipertrigliceridemia Postoperatorio Traumatismo Cáncer Embarazo Insuficiencia cardíaca Síndrome nefrótico Edad Anticonceptivos orales Obesidad + + + +

23
S. HIPERCOAGULABILIDAD HEREDADA
Resistencia a la PCa (FV Leiden) Alteraciones de AT-III Disminución (tipo I) Disfunción (tipo II) Alteraciones de PC (tipo I y II) Alteraciones de PS Aumento de Protombina (G20210A) Alteraciones de la Fibrinolisis Alteración de plasminógeno (disminución y disfunción) Disminución de activadores del Plasminógeno, etc. Hiperhomocisteinemia
 Alteraciones de AT-III. Disminución (tipo I) Disfunción (tipo II) Alteraciones de PC (tipo I y II) Alteraciones de PS. Aumento de Protombina (G20210A) Alteraciones de la Fibrinolisis. Alteración de plasminógeno (disminución y disfunción) Disminución de activadores del Plasminógeno, etc. Hiperhomocisteinemia.")
25
S. HIPERCOAGULABILIDAD HEREDITARIOS Prevalencia y riesgo de trombosis
Población general Trombosis venosa Riesgo relativo de trombosis Deficiencia AT 0.02 25-50 Deficiencia PC 3.0 10-15 Deficiencia PS ? FV Leiden 2-15 20-50 8 PT G20210A 1.0 7-8

26
Tiempo de tromboplastina parcial activada (TTPA)
Activador Ca++

27
Resistencia a la acción de la proteina C activada

28
1. FV-Leiden: Principal causa de SH – 20% de eventos clínicos (AT, PC y PS – 5%) + factor de riesgo PC activado inhibite F Va and F VIIIa. Incapacidad del PCA de inhibir el complejo Va, VIIIa debido a una mutación del FV Heterozigotos: 5-10 veces más rx TE. Homozigotos: veces.

29
Resistencia a la Proteína C Activada (RPCa)
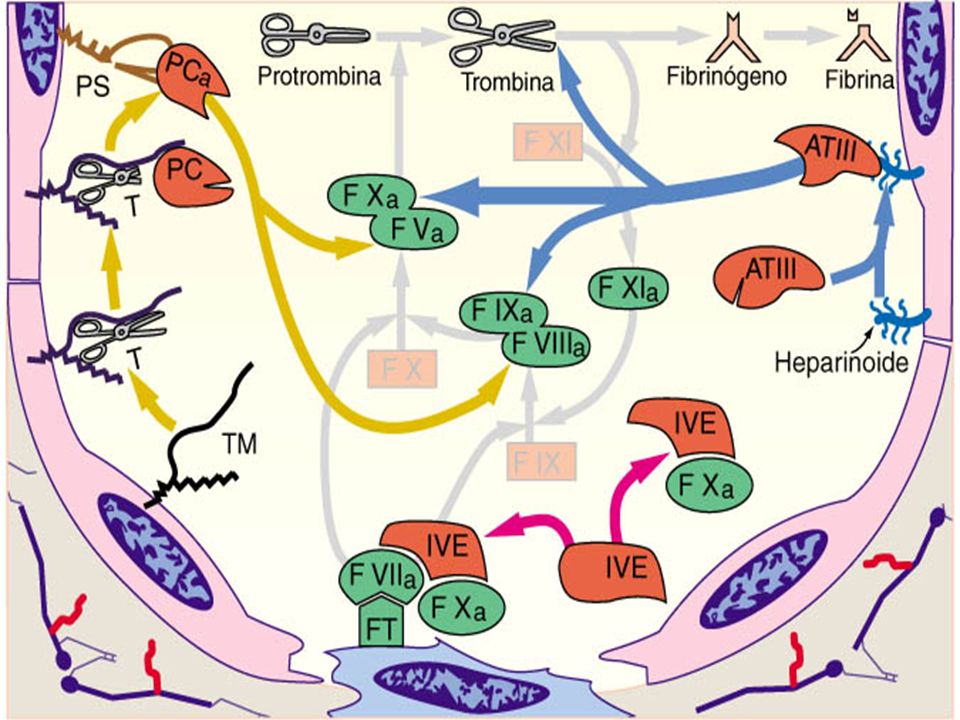
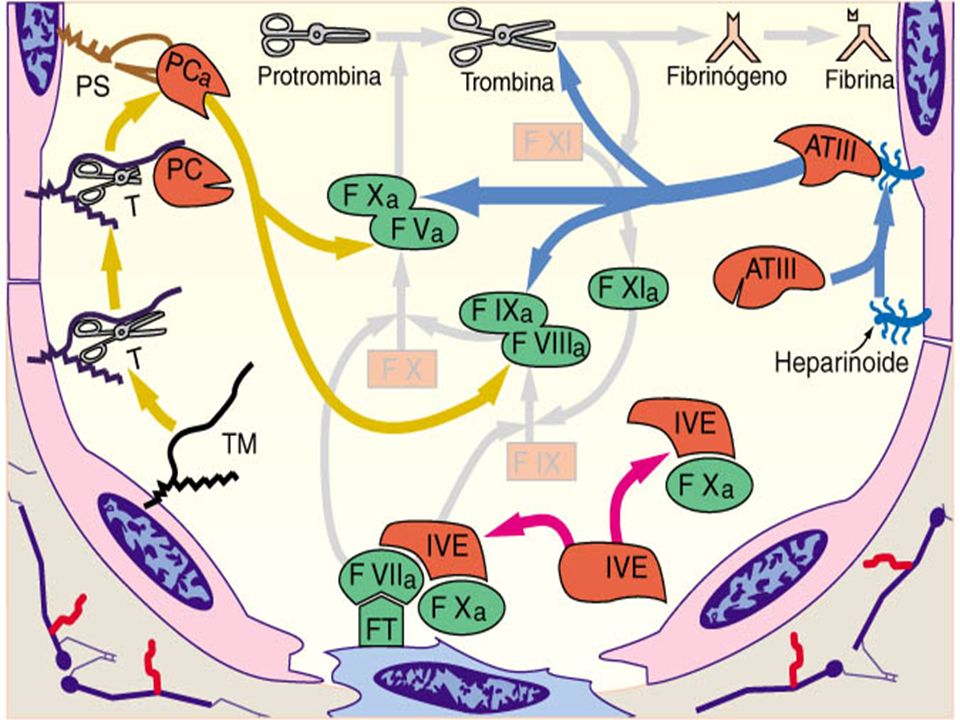
Daño Vascular VI VE XII colágeno XIIa Calicreína PK HMWK Sist. PC FT VII VIIa VIIa-FT VIII XI XIa HMWK Ca+2 IXCa IXa-VIIIa V Ca+2 XIII XIIIa X Xa-Va Protombina Trombina Fibrinógeno Fibrina Fibrina estable Ca+2 Ca+2

30
Resistencia a la Proteína C Activada (RPCa)
20-25% de las trombosis venosas (Casos selecionados) Mutación en el gen del FV (FV Leiden) G1691A A A A C C Pca Trombina FV Leiden: Menos sensible a la degradación por PCa Actividad procoagulante normal Gen FV, crom 1, 25 exones H2N COOH Arg Arg Glu
 Mutación en el gen del FV (FV Leiden) G1691A. A A A C C. Pca Trombina. FV Leiden: Menos sensible a la degradación por PCa. Actividad procoagulante normal. Gen FV, crom 1, 25 exones. H2N. COOH. Arg Arg Glu.")
31
Características principales de la RPCA hereditaria (FV Leiden)
Herencia Autosómica dominante Valores en sujetos afectados Heterocigotos Razón PCA baja Homocigotos Razón PCA muy baja Síntomas trombóticos Trombosis venosa profunda Factores predisponentes Embarazo, cirugía, ACO, etc. Prevalencia Población general % (Chile 1-2%?) Pacientes no seleccionados 4.8% Pacientes seleccionados* % Riesgo relativo de trombosis 8 *<50a, historia familiar, TV recurrente, ausencia de factores adquiridos
 Pacientes no seleccionados 4.8% Pacientes seleccionados* 20.1% Riesgo relativo de trombosis 8. *<50a, historia familiar, TV recurrente, ausencia de factores adquiridos.")
32
Resistencia a la Proteína C Activada (RPCa)
Pruebas de coagulación TTPA paciente + PCa TTPA paciente Paciente pool normal En TAC oral: Plasma del paciente diluido en plasma deficiente en FV Detección de la mutación en el gen del FV (G1691A) Amplificación de segmento génico y estudio con enzima de restricción (MnI I) PCR alelo específica *Valores según Lab. > 2,1* > 0,58*
 Amplificación de segmento génico y estudio con enzima de restricción (MnI I) PCR alelo específica. *Valores según Lab. > 2,1* > 0,58*")
33
Factor V Leiden 300 267 bp 200 bp 200 163 bp 100 67 bp 37 bp G/G
Patrón PM Producto PCR Homo- cigoto A/A Hetero- cigoto G/A Normal G/G 300 267 bp 200 bp 200 163 bp 100 67 bp 37 bp

34
2. Deficiencia de Protein C :
Causa común (aumento TE con la edad). Necesidad de TM de la pared del endotelio . Heterozigotos: 50% del nivel normal Homozigotos: Neonatos nacen con niveles indetectables (trombo microvascular de la piel CID necrosis purpura fulminans).
. Necesidad de TM de la pared del endotelio . Heterozigotos: 50% del nivel normal. Homozigotos: Neonatos nacen con niveles indetectables (trombo microvascular de la piel CID necrosis purpura fulminans).")
35
3. Deficiencia de Protein S :
Co-Factor NO Enzimático para PC Se une al TM-PC. Similares propiedades de PC. Dos formas: Libre en plasma y unida a la proteina C4b (60%). Solo la forma libre es Co-factor de PCA En algunas ocasiones es dificil medir PS Al igual que PC puede ser adquirida: enfermedad hepática, Warfarina, embarazo, cancer, CID y Quimioterapia
. Solo la forma libre es Co-factor de PCA. En algunas ocasiones es dificil medir PS. Al igual que PC puede ser adquirida: enfermedad hepática, Warfarina, embarazo, cancer, CID y Quimioterapia.")
36
4. Deficiencia de Antithrombin III:
Causa común (incidencia 1/2000 – 1/5000; heterozigotos; 50% TVP): Cambios cuantitativos y Cualitativos. (Adquirida: CID, cirrosis). Se une a una trombina inactiva, Factores IXa, Xa, XIa and XIIa (Complejo AT/heparina - frecuencia de inhibición 1000-veces aumentada). No necesariamente un factor de riesgo debe estar presente en heterozigotos para desarrollar TE. Incidencia aumentada conla edad : 80% a los 55 años.
: Cambios cuantitativos y Cualitativos. (Adquirida: CID, cirrosis). Se une a una trombina inactiva, Factores IXa, Xa, XIa and XIIa (Complejo AT/heparina - frecuencia de inhibición 1000-veces aumentada). No necesariamente un factor de riesgo debe estar presente en heterozigotos para desarrollar TE. Incidencia aumentada conla edad : 80% a los 55 años.")
37
Factores inhibidos por el sistema de la ATIII
Daño vascular VI VE XII colágeno XIIa Calicreína PK HMWK VIII FT VII VIIa VIIa-FT XI XIa HMWK Ca+2 IXCa IXa-VIIIa V sist.ATIII Ca+2 XIII XIIIa X Xa-Va Protombina Trombina Fibrinógeno Fibrina Fibrina estable Ca+2 Ca+2

38
Anticoagulantes naturales: disminución o disfunción
Múltiples mutaciones y deleciones descritas ATIII Antígeno 0,8-1,3 U/ml Funcional % PC Antígeno 0,7-1,6 U/ml Funcional % PS Total > 0,58 U/ml Tipo I: Déficit proteína Tipo II: Alteración funcional

39
5. Protrombina anormal (PT G20210 A):
Común. Incrementa los niveles de protrombina aumentando la formación de Trombina Diagnóstico: Técnicas de DNA-PCR .

40
Mutación G20210A del Gen de la Protombina
5´ 3´ Gen de Protombina, 14 exones G20210A Síntesis de Protombina (mayor nivel en plasma; 30%) Búsqueda: PCR y digestión con Enzima de restricción (Hind III) “Nested” PCR Prevalencia: Europa: 1,7- 3% (Pacientes GG:82%, GA:18%, AA:0%) Chile: 1-2.5% Asociación con trombosis venosa y tromboembolismo pulmonar 3-6 veces el riesgo de T. venosa ( asociado a ACO)
 Búsqueda: PCR y digestión con Enzima de restricción (Hind III) Nested PCR. Prevalencia: Europa: 1,7- 3% (Pacientes GG:82%, GA:18%, AA:0%) Chile: 1-2.5% Asociación con trombosis venosa y tromboembolismo pulmonar. 3-6 veces el riesgo de T. venosa ( asociado a ACO)")
41
PT G20210A 300 270 bp 200 148 bp 100 Heterocigoto mutado G/A
Homocigoto mutado A/A Normal G/G 300 270 bp 200 148 bp 100

42
Características principales de la mutación G20210A del gen de la protrombina
Herencia Autosómica dominante Valores en sujetos afectados Heterocigotos % Homocigotos >130% Síntomas trombóticos Trombosis venosa profunda Factores predisponentes Embarazo, cirugía, etc. Prevalencia Población general 1% (Chile 1%) Pacientes no seleccionados 7.1% Pacientes seleccionados* 16% Riesgo relativo de trombosis 3 *<50a, historia familiar, TV recurrente, ausencia de factores adquiridos
 Pacientes no seleccionados 7.1% Pacientes seleccionados* 16% Riesgo relativo de trombosis 3. *<50a, historia familiar, TV recurrente, ausencia de factores adquiridos.")
43
Síndrome de Plaqueta Pegajosa :
Holliday y Mammen 1983: ACV I. Adulto joven Autosómica dominante Formas plaquetas: Redonda o inactiva: 10% Dendrítica o Activación intermedia: 80% Extendida y diseminada o Activación máxima: 10% Estudio actual por agregometría Patologías con Hiperagregabilidad: DM, Síndrome nefrítico, fobrosis quística, anorexia n. Elevación: Factor 4-plaq, TXA2, B-tromboglobulina.

44
Síndrome de Plaqueta Pegajosa :
Especialmente en Trombosis Arterial (IAM, AIT) y recurrencia a pesar del uso de Warfarina. Dx: Hiperagregabilidad: Epinefrina: 11, 1.1, 0.6 mcM/ml ADP: 2.3, 1.2, 0.6 mcM/ml Trasmisión de la luz: 100% agregación 3 Formas: Tipo I: Hiperagregabilidad Epinefrina y ADP Tipo II: Hiperagregabilidad Epinefrina Tipo III: Hiperagregabilidad ADP Si utiliza ASA, debe suspenderla por 14 días previo al examen
 y recurrencia a pesar del uso de Warfarina. Dx: Hiperagregabilidad: Epinefrina: 11, 1.1, 0.6 mcM/ml. ADP: 2.3, 1.2, 0.6 mcM/ml. Trasmisión de la luz: 100% agregación. 3 Formas: Tipo I: Hiperagregabilidad Epinefrina y ADP. Tipo II: Hiperagregabilidad Epinefrina. Tipo III: Hiperagregabilidad ADP. Si utiliza ASA, debe suspenderla por 14 días previo al examen.")
45
Síndrome de Plaqueta Pegajosa :
Dx sugestivo: Hiperagregabilidad (1 conc) e historia de trombosis. Repetir la prueba Dx confirmado: Hiperagregabilidad (2 conc). Repetir la prueba. Hiperagregabilidad (2 conc) e historia de trombosis. Recomendaciones: Pruebas similares en más de una oportunid: Hallazgos deben desaparecer con ASA Hallazgos deben reaparecer cuando se suspenda ASA por 15 días Realizar las demás pruebas de hipercoagulabilidad Pruebas en eventos agudos: Repetir en 4 a 9 meses
 e historia de trombosis. Repetir la prueba. Dx confirmado: Hiperagregabilidad (2 conc). Repetir la prueba. Hiperagregabilidad (2 conc) e historia de trombosis. Recomendaciones: Pruebas similares en más de una oportunid: Hallazgos deben desaparecer con ASA. Hallazgos deben reaparecer cuando se suspenda ASA por 15 días. Realizar las demás pruebas de hipercoagulabilidad. Pruebas en eventos agudos: Repetir en 4 a 9 meses.")
46
Adhesión y agregación plaquetaria Tratamiento
ASA Endotelio vascular PGI2 Inductor de agregación Ac. Araquidónico PGG2 Tromboxano A2 TXA2 Fosoflipasa Cicloxigenasa ATP AMPc AMP Adenil ciclasa + Ca++ Membrana Plaquetar Cuerpos densos Trpmboxano sintetasa GPIIb-IIIa Fibrinógeno Plaqueta GPIb-IX FvW GPIa-IIa Endotelio dañado

47
Polimorfismos de las glicoproteínas de membrana plaquetaria, como factor de riesgo de Enf. coronaria
Plaquetas contribuyen al trombo arterial Adhesión mediada por proteína adhesivas (GP) GP plaquetarios son polimórficas Sistemas antigénicos (HPA-1, -6) Asociación con trombosis arterial I. miocardio Normales GPIa C807T , Santoso S y cols GPIIIa HPA-1b % % p 0,01 Pereira J y cols GPIb VNTR* D Murata M y cols * 4 isoformas: A-D; D más grande; T: número de GPIa
 GP plaquetarios son polimórficas. Sistemas antigénicos (HPA-1, -6) Asociación con trombosis arterial. I. miocardio Normales. GPIa C807T 1,5-2 Santoso S y cols. GPIIIa HPA-1b 22% 12% p 0,01 Pereira J y cols. GPIb VNTR* D Murata M y cols. * 4 isoformas: A-D; D más grande; T: número de GPIa.")
48
Homocisteína (Hcy) Aminoácido tiól No esencial
Intermediario en el metabolismo de la Metionina

49
(MTHFR)
")
50
Hiperhomocisteinemia y Trombosis
Daño directo del endotelio Disminución de expresión de la Trombomodulina Disminución de actividad de la PC Aumento de actividad de FV Aumento de la actividad plaquetaria

51
Homocisteína Plasmática (pHcy)
Aumenta Hombres, Edad, Tabaquismo, Café, Alcohol (exceso) Defectos genéticos del metabolismo Medición Cromatografía líquida de alta presión (HPLC), ELISA Homocisteína total (8,8 ± 0,3 µM/L) Tratamiento Acido fólico Homocisteína Acido fólico
 Defectos genéticos del metabolismo. Medición. Cromatografía líquida de alta presión (HPLC), ELISA. Homocisteína total (8,8 ± 0,3 µM/L) Tratamiento. Acido fólico. Homocisteína. Acido fólico.")
52
5,10 Metilentetrahidrofolato Reductasa (MTHFR)
Mutación C677T Ala Val (PCR y enzima de restricción) MTHFR termolábil de Homocisteína plasmática (pHcy) Factor de riesgo de Trombosis
 MTHFR termolábil. de Homocisteína. plasmática (pHcy) Factor de riesgo de Trombosis.")
53
Hiperfibrinogenemia

54
Factores de riesgo se potencian
Ejemplos Riesgo relativo de Factor de riesgo trombosis arterial FV Leiden 2,5 Obesidad 3,7 FV Leiden + Obesidad 19,3

55
S. HIPERCOAGULABILIDAD
Hereditarias Adquiridas

56
S. HIPERCOAGULABILIDAD ADQUIRIDA
Condiciones predisponentes Cirugía, trauma, Inmovilización prolongada Edad avanzada Uso de anticonceptivos orales Embarazo, puerperio Terapia de reemplazo hormonal Cáncer, enfermedades mieloproliferativas

57
S. HIPERCOAGULABILIDAD ADQUIRIDO
Ac antifosfolípidos (aFL) Ac Inducidos por heparina (AcIH)
 Ac Inducidos por heparina (AcIH)")
58
CLASIFICACIÓN DEL SAF Primario Secundario Enf.Auntoinmunes LES
Variantes del SAF SAF catastrófico Asociado a otros Sínd.microangiopáticos Síndrome de hipoprotrombinemia SAF seronegativo Primario Secundario Enf.Auntoinmunes LES Lupus probable Otras mesenquimopatías Neoplasias Tumores sólidos Neoplasias hematológicas Inducido por drogas Asociado a infección

59
Antígenos Beta-2 Glicoproteína I (b2GPI) Protrombina (PT)
Proteínas del Sistema Proteína C PC, PS, Trombomodulina Anexina-V Kininógeno de alto peso molecular Fosfolípidos aniónicos: FS, CL Antígenos Beta-2 Glicoproteína I (b2GPI) Protrombina (PT)
 Protrombina (PT)")
60
b2GPI Patrón de PM b2GPI kDa I II III 29 IV V NH2 88 200 97 68 50 43
+ III IV V II I NH2 N k E 288 281 C 88 247 306 316 Patrón de PM b2GPI kDa 200 97 68 50 43 29

61
Mecanismos Trombogénicos de los Ac aFL
Monocitos Aumento expresión de FT en monocitos circ. Mecanismos Trombogénicos de los Ac aFL Inhibición de anticoagulantes naturales Sistema PC Anexina V Activación celular Plaquetas Liberación de Micropartículas Aumento TXA2 Células endoteliales Desbalance TXA2 / PGI2 Up regulation de Moléculas de Adhesión Aumento expresión FT Aumento unión de Protrombina

62
Pruebas Diagnósticas ELISAs de fase sólida Anticoagulante Lúpico
Citometría de flujo

63
aCL clásico Unidad Leve Moderado Alto aCL IgG GPL 12-39 40-80 >80
>80 aCL IgM MPL Asociación clínica de aCL IgG moderado o alto IgM e IgA menos frecuente Persistente

64
Anticoagulante Lúpico (AL)
Prolongación de Pruebas de Coagulación dTTI Inhibición de Tromboplastina Tisular diluida KCT Tiempo de Coagulación con Kaolin dRVVT Tiempo de Veneno de Vívora de Russell Pruebas para diagnóstico

65
Algoritmo de AL* AL * Subgrupo de aCL y aPT Normal Deficit de
Prueba de Coagulación (dTTI, KCT, dRVVT) Normal Anormal Prueba coagulación Paciente+Normal Normaliza No normaliza Deficit de factor(es) Neutralización (con FL plaq.) AL Otro Inhibidor * Subgrupo de aCL y aPT
 Normal. Anormal. Prueba. coagulación. Paciente+Normal. Normaliza. No. normaliza. Deficit de. factor(es) Neutralización. (con FL plaq.) AL. Otro. Inhibidor. * Subgrupo de aCL y aPT.")
66
S. HIPERCOAGULABILIDAD HEREDITARIOS Causas
Frecuentes Factor V Leiden Mutación G20210A del gen de la protrombina Homocigocidad para mutación C677T de la MTHFR Raras Deficiencia de antitrombina Deficiencia de proteína C Deficiencia de proteína S

67
INACTIVACION DEL FACTOR Va POR PCA
1 306 NH2 A1 A2 COOH Ca2+ 2183 NH2 A3 C1 C2 COOH PCA 1 306 Arg 506 NH2 A1 COOH Ca2+ 2183 NH2 A3 C1 C2 COOH

68
Factor V Leiden L D R R R I Q CTG GAC AGC CGA GGA ATA CAG
PCA L D R R R I Q CTG GAC AGC CGA GGA ATA CAG CTG GAC AGC CAA GGA ATA CAG L D R Q R I Q 506 1.691 PCA

70
Estudio de laboratorio de trombofilia ¿Qué pruebas?
Ensayo funcional (actividad cofactor heparina anti-Xa) Ensayo funcional (cromogénico con veneno de serpiente) Ensayo funcional (inespecífico) Antígeno total o libre Método basado en TTPA con o sin plasma deficiente en FV. Confirmación de resultados (+) con genotipo de FV. Genotipo Pruebas dependientes de FL para ACL (KCT y dRVVT) y anticuerpos anticardiolipinas HPLC, inmunoensayo Antitrombina Proteína C Proteína S Resistencia a la PCA Mutación G20210A de PT Anticuerpos antifosfolípidos Homocisteína
 Ensayo funcional (cromogénico con veneno de serpiente) Ensayo funcional (inespecífico) Antígeno total o libre. Método basado en TTPA con o sin plasma deficiente en FV. Confirmación de resultados (+) con genotipo de FV. Genotipo. Pruebas dependientes de FL para ACL (KCT y dRVVT) y anticuerpos anticardiolipinas. HPLC, inmunoensayo. Antitrombina. Proteína C. Proteína S. Resistencia a la PCA. Mutación G20210A de PT. Anticuerpos antifosfolípidos. Homocisteína.")
71
Quienes estudiar: Jóvenes: < 50 años, TE recurrente, sitio inusual, TE con Warfarina. No estudialr en el episodio agudo (inhibidores de la coagulación pueden estar disminuidos). Ideal: Estudiar 6 semanas después del EA. Pacientes con Warfarina: PC & PS son Vit K dependentes, pueden estar bajos falsamente).
. Ideal: Estudiar 6 semanas después del EA. Pacientes con Warfarina: PC & PS son Vit K dependentes, pueden estar bajos falsamente).")
72
GRACIAS

Presentaciones similares