Descargar la presentación
La descarga está en progreso. Por favor, espere

1
Errores congénitos del metabolismo
Dra.Alejandra Acosta Gualandri Hospital Nacional de Niños Servicio de Genética

2
Definiciones Error: Congénito: Metabolismo:
defecto (mutación que altera la expresión fenotípica) Congénito: innato Metabolismo: bioquímica que produce acumulación metabolito o deficiencia de sustrato
 Congénito: innato. Metabolismo: bioquímica que produce acumulación metabolito o deficiencia de sustrato.")
3
Metabolismo es la suma de todas las reacciones químicas que ocurren como parte del proceso continuo de renovación y eliminación en el cuerpo: Conversión Producción Transporte 3

4
Se conocen más de 500 errores congénitos del metabolismo hoy en día.
Poco frecuentes (1/600), con alta morbi-mortalidad Puede verse afectado: Enzima Hormona Receptor Transportador Elemento estructural Vias metabólicas involucradas: Proteinas Carbohidratos Lipidos
, con alta morbi-mortalidad. Puede verse afectado: Enzima. Hormona. Receptor. Transportador. Elemento estructural. Vias metabólicas involucradas: Proteinas. Carbohidratos. Lipidos.")
5
A B C

6
ECM se pueden manifestar de 2 maneras:
Enfermedades que involucran un solo órgano o sistema. 2. Enfermedades en las cuales el defecto bioquímico de base, afecta un paso metabólico común de multiples células u órganos o afecta a un órgano que involucra reacciones sistémicas y humorales. Alteración metabolismo de moléculas complejas (deposito) Alteración del metabolismo intermedio (intoxicación) Defectos de la producción de energia
 Alteración del metabolismo intermedio. (intoxicación) Defectos de la producción de energia.")
7
Acumulación de sustrato

8
Producto deficiente

9
Historia clinica es muy importante:
En fase aguda las emergencias metabólicas y endocrinológicas arriesgan la vida del paciente. Llegan a ER sin un Dx, debido a que el estress o infecciones desencadenan su aparición. Historia clinica es muy importante: Enfermedades recientes, infecciones, traumas Estado neurológico previo y actual Historia familiar de abortos o muertes tempranas Consanguinidad (sin embargo mayoría de la enf son AR) Dx anterior en hermano 9
 Dx anterior en hermano. 9.")
10
Presentación tardía no debe excluir el Dx de enf metabólica.
Intervalo desde el nacimiento hasta la presentación de síntomas: horas a meses. Presentación tardía no debe excluir el Dx de enf metabólica. Sx neurológico representan el 85%, seguido por sx digestivos en 58,5% Efecto de la acumulación de metabolitos tóxicos en el SNC Durante embarazo cruzan la placenta Eliminación por la madre durante gestación 10

11
Un EIM se debe considerar:
RN con enfermedad inexplicable, abrupta y progresiva, con historia de un embarazo y parto normal. En todo niño con deterioro agudo y/o alteración conciencia con historia de vomitos, fiebre o hipoglicemia. En todo niño con sintomas y signos de hipoglicemia o acidosis sin causa aparente.

12
Manifestaciones clínicas de ECM
Alteraciones neurológicas/sensorio Retraso del desarrollo psicomotor Convulsiones Hipotonía, Tono muscular anormal Rechazo alimentos Defectos ciclo urea Aversión a frutas y dulces en la intol a fructuosa Encefalopatía metabólica aguda Acidemias orgánicas Defectos del ciclo de la urea Defectos de los metabolismos de amino ácidos

13
Manifestaciones clínicas de ECM
Letargia/Coma Coma hiperamonemico Hemorragia intracraneal y edema cerebral Apnea, distress respiratorio Apnea de origen central Hiperventilación de origen central Defectos del ciclo urea Vómitos Intolerancia a la proteína (cirugía por sospecha errónea de E. Pilorica)
")
14
Manifestaciones clínicas de ECM
Disfunción hepática Coagulopatia/sangrado Hepatomegalia Ictericia Descompensación cardiáca Disfunción muscular Rabdomiolisis Sospecha de sepsis neonatal (inicio rápido) Hipoglicemia Acidosis
 Hipoglicemia. Acidosis.")
15
Title 15

16
Title Title 16

18
DIFERENCIA ENTRE ENFERMEDAD DE ORGANELAS Y DE MOLECULAS PEQUEÑAS
CARACTERISTICA Presentación Evolución Hallazgos físicos Histopatología Respuesta a tx ENF. ORGANELA Gradual Progresiva Características Cambios esp. Pobre ENF. MOL PEQ. Súbito Intermitente Inespecífico

19
OLORES INUSUALES DE ORINA
ENFERMEDAD OLOR Orina del Jarabe de Arce Miel de maple, azúcar quemado Acidemia Isovalérica Queso, sudor de pies Deficiencia múltiple de carboxilasa Orina de gato Fenilcetonuria Moho, ratón mojado Hipermetionemia Mantequilla rancia, repollo podrido Trimetilaminuria, Tirosinemia Pescado descompuesto

20
HALLAZGOS CLINICOS ASOCIADOS A ECM
Enfermedad Hepatomegalia GSD, Def. fructosa 1-6 difosfatasa Hepatoesplenomegalia Gaucher, N. Pick, Sialidosis Macrocefalia Canavan, Gangliosidosis Microcefalia T. de electrones, Leigh Cara grotesca Sialidosis, MPS Macroglosia Pompe, GM1 Distonía o signos extrapiramidales Gaucher, Krabbe, PKU, Lesch Nyhan Manchas rojo cereza N. Pick, Tay-Sachs Retinitis pigmentaria Enf. mitocondrial, Zellweger Atrofia ótica Zellweger, Piruvato Deshidrogenasa Opacidad corneal I cell, MPS, Fabry

21
HALLAZGOS CLINICOS ASOCIADOS A ECM
Enfermedad Cataratas Galactosemia, Enf. Peroxisomas Dislocación del cristalino Def. Sulfito Oxidasa, Homocistinuria Anomalías óseas Peroxisomas, Lisosomas Piel gruesa Sialidosis, MPS, I Cell Nódulos cutáneos Def. Ceramidasa, Def. Lipoproteinlipasa Ictiosis Gaucher, Def. Esteroide sulfatasa Alopecia Def. Carboxilasa múltiple, Menkes Descamación de piel A. Propionica, Porfiria Pelo ensortijado Menkes Ictericia Trast. de Ac. Biliares, Wilson, Galactosemia Crisis neurológicas Ciclo de la urea, Acidemias orgánicas

22
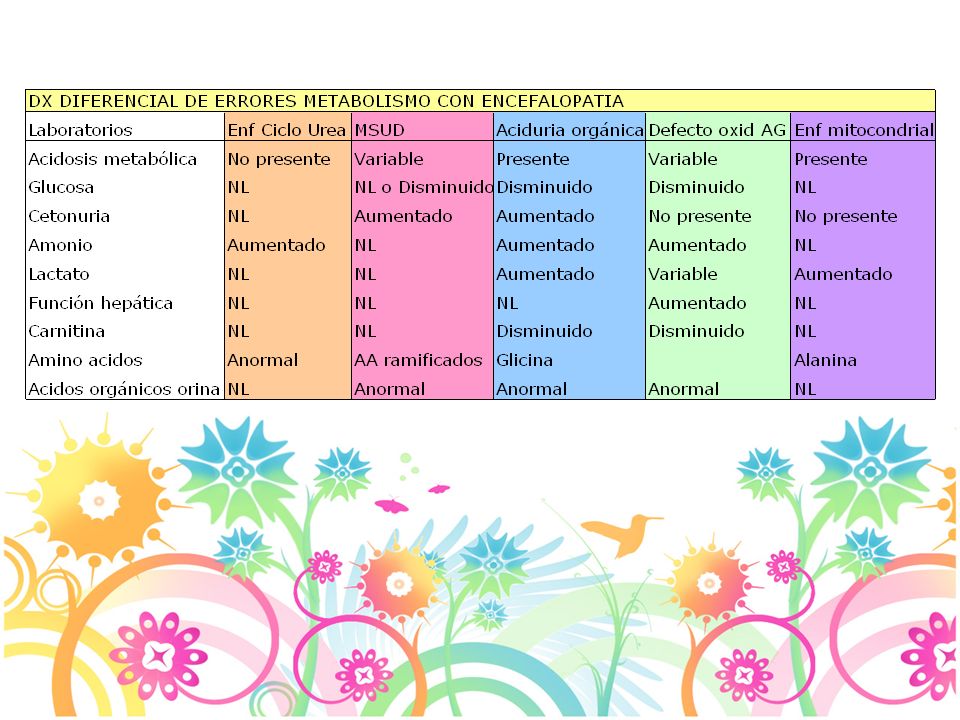
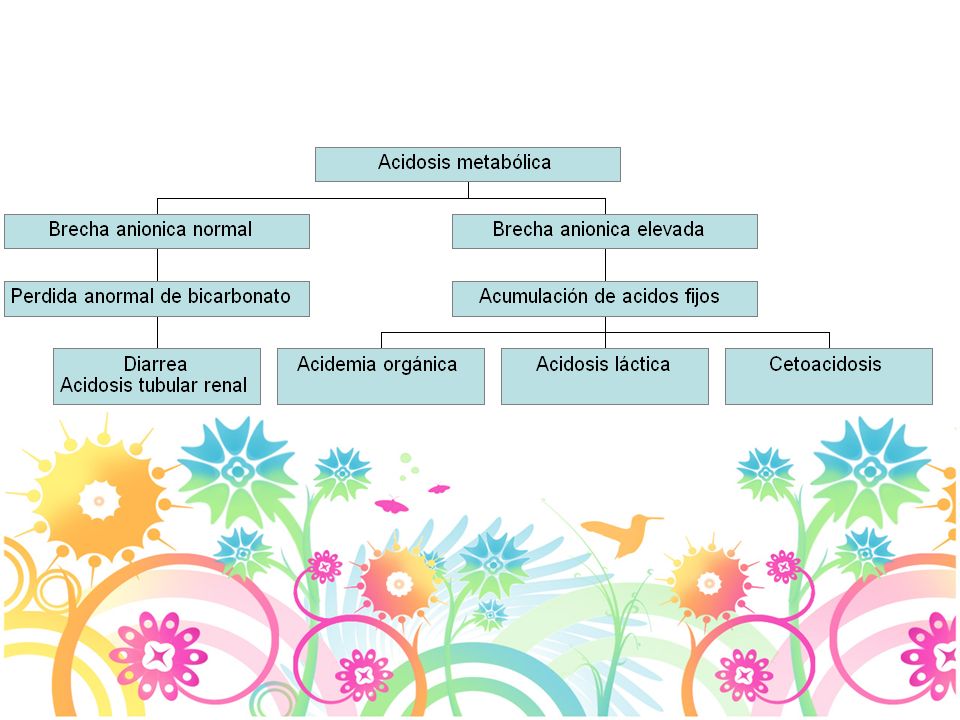
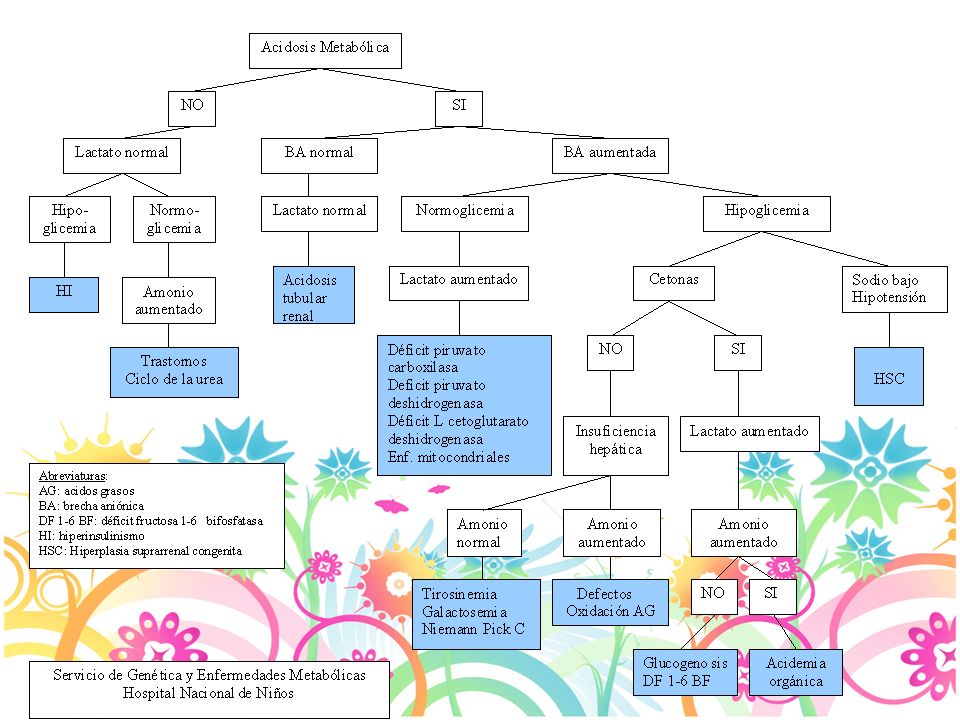
Diagnóstico Acidosis metabólica Hiperamonemia Acidosis láctica
Hipoglicemia Enfermedad cardiaca: Mucopolisacaridosis, Pompe, Mitocondriales Disfunción hepática Dismorfismo: mucopolisacaridosis, Zellweger 22

23
Diagnóstico Acidosis metabólica pH normal: 7.35-7.45
Alcalemia: pH > Acidemia: pH <7.35 Bicarbonato: mEq/L Alcalosis: HCO3 elevado, pH nl Acidosis: HCO3 disminuido, pH nl PaCo2: mm Hg Pa02: mm Hg Brecha anionica: Na - (Cl + HCO3)= 14 +/-2 Calcular el exceso de Brecha anionica BA pte - BA normal(12)+ HCO3 > 30 mmol/L alcalosis metabólica de fondo < 25 mmol/L acidosis metabólica de fondo 23
= 14 +/-2. Calcular el exceso de Brecha anionica. BA pte - BA normal(12)+ HCO3. > 30 mmol/L alcalosis metabólica de fondo. < 25 mmol/L acidosis metabólica de fondo. 23.")
25
Concentración de glucosa:
1 mmol/L= 18 mg/dl 10 mg/dl= 0.55 mmol/L Hipoglicemia Glicemias < 40 mg/dl en el RN Causas más frecuentes: hiperinsulinismo o hipopituitarismo Cetonas: Elevadas: hipoglicemia cetósica, ac.orgánicas, EDG Normales: hiperinsulinismo, hipopituitarismo, defecto oxid AG Lactato: Elevado con hepatomegalia: EDG Elevado sin hepatomegalia: Ac orgánicas, defecto oxid AG Disfxn hepática: defecto oxidación AG, tirosinemia, intol a fructosa 25

26
Tx de Hipoglicemia Tx: AG:7-10 mg/kg/min, SG 10%
Mantener glicemias 100mg/dl Si se necesitan bolos de glucosa no más de 200mg/kg (glucosa 20% 1 cc/kg o SG 10% 2cc/kg 26

27
Concentración amonio:
Hiperamonemia Amonio: Normal RN < 110 umol/L Enfermo RN hasta 180 umol/L EIM >200 umol/L Luego periodo RN > 100umol/L Amonio elevado: Desordenes ciclo urea Acidemias orgánicas Falla hepática Hiperamonemia transitoria por Qx cardiaca Aumento de activ muscular (convulsiones, VMA) Concentración amonio: umol/L= ug/dl x 0.59 27
 Concentración amonio: umol/L= ug/dl x")
28
Tx de Hiperamonemia Suspender ingesta de proteina.
Remover exceso amonio PRONTO con valores > 500umol/L hemofiltración, hemodialisis, dialisis peritoneal Aporte de glucosa con SG 10% y electrolitos Suplementar intermediarios del ciclo de la urea con arginina o citrulina Arginina 360 mg/kg (2 mmol/kg= 2ml/kg de sol. 1 Molar) Benzoato de sodio: mg/kg/dia VO Permitir el metabolismo mitocondrial con L-carnitina L- Carnitina 30%: mg/kg (1cc= 300 mg), VO Excreción renal de amonio: dar liquidos, forzar diuresis. 28
 Benzoato de sodio: mg/kg/dia VO. Permitir el metabolismo mitocondrial con L-carnitina. L- Carnitina 30%: mg/kg (1cc= 300 mg), VO. Excreción renal de amonio: dar liquidos, forzar diuresis. 28.")
29
Concentración lactato:
mmol/L= mg/dl x 0.11 Lactato elevado Normal en sangre: < 2.1 mmol/L, <20 mg/dl Normal en LCR: < 1.8 mmol/L, < 16 mg/dl Elevado en: Uso de torniquete o dificultad toma muestra Activ muscular excesiva, convulsiones Hipoxia central o periferica Isquemia, shock, falla cardiaca, Insuf renal y hepática Sepsis EIM: defectos cadenas resp, deficit piruvato quinasa, defecto oxid AG, EDG, Ac orgánicas 29

30
Tx de Lactato elevado Adecuada ingesta calórica, fluidos y electrolitos. Evitar las condiciones con alto consumo de energia Fiebre/convulsiones L-carnitina 30% a mg/kg/dia, VO en tres dosis Evitar drogas que inhiben cadena respiratoria: valproato, tetraciclina, cloranfenicol 30

33
Tx de emergencia 1. Eliminar la ingesta y remover el metabolito acumulado Eliminación del tóxico (liquidos,forzar diuresis, hemodiálisis, hemofiltración o dialisis peritoneal) Intolerancia a proteína se debe descontinuar proteina 2. Iniciar manejo farmacológico y vitamínicos específico Defectos del ciclo de la urea con hiperamonemia: Benzoato de sodio, Fenilbutirato: quelantes de amonio L- Arginina: permite la ureagenesis 33
 Intolerancia a proteína se debe descontinuar proteina. 2. Iniciar manejo farmacológico y vitamínicos específico. Defectos del ciclo de la urea con hiperamonemia: Benzoato de sodio, Fenilbutirato: quelantes de amonio. L- Arginina: permite la ureagenesis. 33.")
34
Tx de emergencia 3. Acidosis metabólica Bicarbonato IV
Vitamina B12 a 1 mg IM en formas de acidemia metilmalonica con respuesta a B12. Biotina 10 mg/dia por VO o SNG en deficiencias de carboxilasa múltiple y acidemia propiónica 3. Acidosis metabólica Bicarbonato IV producción ácidos es constante por lo que es difícil calcular déficit Monitoreo de condición acido base 34

35
Tx de emergencia 4. Prevenir en catabolismo:
Alimentación enteral o parenteral Proveer aporte calórica mínimo de cal/kg/dia, sea IV o VO Administración glucosa IV, > aporte calórico posible Lípidos IV en defectos ciclo urea o defectos donde las grasas no jueguen rol importante 5. Iniciar proteínas a los 2- 3 dias de restricción proteica completa AA esenciales VO o IV, dosis inicial de 0.5 g kg/dia hasta llegar a 1 g kg/dia, mantener hasta Dx definitivo. 35

36
Tx de emergencia 6. Ajustar liquidos, calorias y requerimientos minimos de forma individual usando como parametros el crecimiento del paciente y los niveles plasmáticos del agente agresor. 36

37
MUCHAS GRACIAS !!

Presentaciones similares




>")















