Descargar la presentación
La descarga está en progreso. Por favor, espere

1
Dra. María Sofía Giménez
Lipidosis Tema 16 (Bolilla5) Dra. María Sofía Giménez
 Dra. María Sofía Giménez.")
2
TEMA 16: Lipidosis. Esfingoglucolípidos. Deficiencia de ceramidasa
TEMA 16: Lipidosis. Esfingoglucolípidos. Deficiencia de ceramidasa. Enfermedad de Niemann-Pick. Enfermedad de Gaucher. Leucodistrofia globoide y metacromática. Deficiencia múltiple de sulfatasa. Enfermedad de Fabry. Gangliosidosis Enfermedad de Wolman. Diagnóstico. Enzimoterapia. Diagnóstico prenatal. Participación en la transducción de señales.

3
TRASTORNO DEL METABOLISMO DE LOS LÍPIDOS

4
Trastornos del metabolismo de los lípidos
CLASIFICACIÓN de los lípidos Simples o triglicéridos Complejos : esfingolípidos, fosfolípidos y colesterol.

5
Trastornos del metabolismo de los lípidos. Clasificación
1.- Trastornos de la oxidación mitocondrial de los ácidos grasos 2.-Trastornos de los ácidos grasos de cadena muy larga ó trastornos peroxisomales: Adrenoleucodistrofia ligada al cromosoma X Adrenoleucodistrofia autosómico-recesiva Síndrome de Zellweger (cerebro-hepato-renal) 3.- Lipidosis 4.- Trastornos del metabolismo y transporte de las lipoproteinas
 3.- Lipidosis. 4.- Trastornos del metabolismo y transporte de las lipoproteinas.")
6
Metabolismo de A. Grasos
Metabolismo AG en mitocondrias y peroxisomas. Importante fuente de energía, sobre todo en situaciones de ayuno y estrés metabólico. Corazón, músculo esquelético e hígado, órganos particularmente dependientes de esta vía (cuerpos cetónicos). Más de 25 enzimas y transportadores implicadas en esta vía: Etapas: Captación y Activación de los AG por las células. Ciclo de la Carnitina Espiral de la betaoxidación. Síntesis de los Cuerpos Cetónicos. El producto final es el Acetil-coA.
. Más de 25 enzimas y transportadores implicadas en esta vía: Etapas: Captación y Activación de los AG por las células. Ciclo de la Carnitina. Espiral de la betaoxidación. Síntesis de los Cuerpos Cetónicos. El producto final es el Acetil-coA.")
7
Metabolismo de los ácidos grasos

8
Los trastornos de la β-Oxidación : grupo complejo de enfermedades:
Trastornos de la beta-oxidación mitocondrial de los ácidos grasos (MCAD) Los trastornos de la β-Oxidación : grupo complejo de enfermedades: >25 entidades diferentes. • Base genética: Incidencia subestimada. Espectro clínico y pronóstico extremadamente variable. Trastorno más frecuente: Déficit de MCAD.
 Los trastornos de la β-Oxidación : grupo complejo de enfermedades: >25 entidades diferentes. • Base genética: Incidencia subestimada. Espectro clínico y pronóstico extremadamente variable. Trastorno más frecuente: Déficit de MCAD.")
9
Trastornos de la beta-oxidación mitocondrial de los ácidos grasos (MCAD): Clínica
Hipoglucemia en ayuno, habitualmente sin cetonuria Miocardiopatía y/o miopatía de gravedad variable Hepatopatía de gravedad variable Otros: Muerte súbita Tubulopatía Polineuropatía

10
Trastornos de la beta-oxidación mitocondrial de los ácidos grasos (MCAD): Diagnóstico
• Sospecha clínica!!! • Demostración de metabolitos anómalos en plasma o en orina. • Demostración del déficit enzimático en cultivo de fibroblastos. • Demostración de la mutación: Genética molecular

11
Trastornos de la beta-oxidación mitocondrial de los ácidos grasos (MCAD)
Tratamiento • Evitar periodos de ayuno. • Controlar la lipolisis mediante una dieta rica en carbohidratos de absorción lenta. • Limitar ingesta de los AG que se vean alterados en función del trastorno metabólico. • Suplementos con Carnitina: Controvertido. • Riboflavina: Controvertido.

12
Adrenoleucodistrofia
Forma Neonatal: Autosómica recesiva Forma Infantil: Recesiva ligada a X

13
Adrenoleucodistrofia

14
Adrenoleucodistrofia
Forma Neonatal: Hipotonía severa progresiva Muerte precoz. Forma Infantil: Leucoencefalopatía progresiva: Espasticidad Insuficiencia suprarrenal

15
Adrenoleucodistrofia Infantil

16
Adrenoleucodistrofia Infantil

17
Adrenoleucodistrofia: Diagnóstico
Sospecha Clínica Elevación de Ácidos Grasos de Cadena Muy Larga Resonancia Magnética Nuclear Confirmación: Posibilidad de estudio genético

19
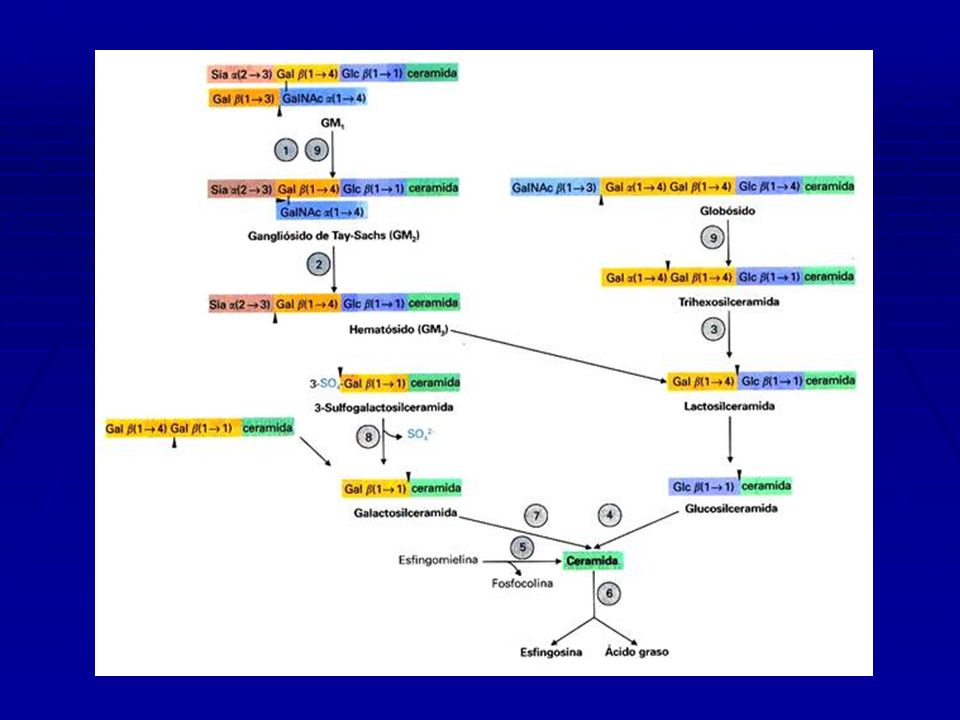
LIPIDOSIS Depósito de lípidos en los lisosomas en forma de glucoesfingolípidos Déficit de una hidrolasa específica Manifestaciones clínicas por depósito en SNC y /o tejidos periféricos Los esfingolípidos forman parte de la membrana celular. Su estructura básica es la esfingosina

20
LIPIDOSIS Esfingosina Aminoácido serina + Ácido palmítico
Esfingosina + A. Graso = Ceramida Glucocerebrósido Esfingomielina Galactocerebrósido Gangliósido Son compuestos que deben estar degradados o reciclados por las enzimas lisosomales

23
LIPIDOSIS 1.- Gangliosidosis GM1
2.- Enfermedad de Tay-Sachs (gangliosidosis GM2 I) 3.-Enfermedad de Sandhoff (gangliosidosis GM2 II) 4.-Enfermedad de Niemann-Pick 5.-Enfermedad de Gaucher 6.-Enfermedad de Fabry
 3.-Enfermedad de Sandhoff (gangliosidosis GM2 II) 4.-Enfermedad de Niemann-Pick. 5.-Enfermedad de Gaucher. 6.-Enfermedad de Fabry.")
24
LIPIDOSIS 7.- Enfermedad de Schindler 8.- Leucodistrofia metacromatica
9.- Deficit de sulfatasa multiple 10.-Enfermedad de Krabbe 11.- Enfermedad de Batten 12.-Enfermedad de Wolman 13.-Fucosidosis

25
GANGLIOSIDOSIS GM1 Déficit de la enzima lisosomal beta-galactosidasa GM1 presente en sustancia gris y blanca cerebral y en los tejidos periféricos Genética: brazo corto del cromosoma 3 Forma infantil y tardía

27
GANGLIOSIDOSIS GM1 Forma infantil
- Hepatoesplenomegalia al nacer Edema de extremidades Erupciones cutáneas neonatales Retraso psicomotor y convulsiones precoces Mancha color rojo cereza en la mácula( 50%) Hernias y edema escrotal Facies tosca, macroglosia cifosis lumbar, rigidez articular (Fenotipo= enfermedad de Hurler).
 Hernias y edema escrotal. Facies tosca, macroglosia cifosis lumbar, rigidez articular (Fenotipo= enfermedad de Hurler).")
28
GM1

29
GANGLIOSIDOSIS GMI Forma Infantil
Miocardiopatía: Cardiomegalia e hipertrofia ventricular izquierda. Alteraciones radiológicas : Disóstosis múltiple (similares a las de las mucopolisacaridosis) TAC y RM cerebral : Ventriculomegalia y atrofia cerebral generalizada - Retraso psicomotor y convulsiones precoces - Muerte precoz a los 3-4 años con disfagia, sordera y ceguera
 TAC y RM cerebral : Ventriculomegalia y atrofia cerebral generalizada. - Retraso psicomotor y convulsiones precoces. - Muerte precoz a los 3-4 años con disfagia, sordera y ceguera.")
30
Fenotipo

31
GANGLIOSIDOSIS GM1 Forma Tardía
Edad de aparición variable Ataxia, disartria y espasticidad como en la parálisis cerebral No afectación visceral No facies tosca No disóstosis multiple Deterioro clínico lento Sobreviven hasta la cuarta década de vida

32
GANGLIOSIDOSIS GMI DIAGNÓSTICO Clínica
Médula ósea, biopsia pulmonar y hepática = células espumosas Queratán sulfato en hígado y orina Estudio enzimático de los leucocitos ó de fibroblastos cutáneos Diagnóstico prenatal TRATAMIENTO - Cuidados sintomáticos

33
ENFERMEDAD DE TAY-SACHS
Afecta al SNC Frecuente en judíos ashkenazi (1/4000) Déficit de beta-hexosaminidasa A y B A los 5 meses hiperacusia + reducción del contacto ocular, enfoque visual y ceguera Retraso psicomotor con hipotonía grave al final del primer año de vida y convulsiones Aumento tamaño craneal sin hidrocefalia No alteraciones óseas Muerte entre los 2-4 años Forma tardía ó juvenil: ataxia y disartria
 Déficit de beta-hexosaminidasa A y B. A los 5 meses hiperacusia + reducción del contacto ocular, enfoque visual y ceguera. Retraso psicomotor con hipotonía grave al final del primer año de vida y convulsiones. Aumento tamaño craneal sin hidrocefalia. No alteraciones óseas. Muerte entre los 2-4 años. Forma tardía ó juvenil: ataxia y disartria.")
34
TAY-SACHS

35
TAY-SACHS

36
Herencia Tay-Sachs

37
Enfermedad de Tay-Sachs

38
ENFERMEDAD DE TAY-SACHS
DIAGNOSTICO Clínica : retraso grave+ mancha rojo cereza, no hepatoesplenomegalia Déficit de beta-hexosaminidasa A en plasma, cultivo de fibroblastos cutáneos ó leucocitos Genética molecular : cromosoma 15 Diagnóstico prenatal ( 1 de cada 30 son portadores) TRATAMIENTO No existe
 TRATAMIENTO. No existe.")
39
ENFERMEDAD DE NIEMANN-PICK
Judíos de origen ashkenazi Déficit de esfingomielinasa ( tipo Ay B) Inicio a los 3-4 meses Fallo de medro Retraso psicomotor y deterioro neurológico progresivo Hepatoesplenomegalia No disóstosis multiple Tipo A mancha rojo cereza ( 50%) Tipo B no hay mancha rojo cereza
 Inicio a los 3-4 meses. Fallo de medro. Retraso psicomotor y deterioro neurológico progresivo. Hepatoesplenomegalia. No disóstosis multiple. Tipo A mancha rojo cereza ( 50%) Tipo B no hay mancha rojo cereza.")
40
ENFERMEDAD DE NIEMANN-PICK
DIAGNÓSTICO Células espumosas en la médula osea Genética molecular: cromosoma 11 TRATAMIENTO No existe

41
Enfermedad de Niemann-Pick

42
ENFERMEDAD DE GAUCHER Depósito de glucosilceramida
( glucocerebrósido ) en el sistema reticuloendotelial La forma clásica es frecuente en los judíos ashkenazi ( 1 de cada 500) y no afecta al SNC Afectación neurológica variable según tipo Forma juvenil asociada a deterioro neurológico de inicio tardío
 en el sistema reticuloendotelial. La forma clásica es frecuente en los judíos ashkenazi ( 1 de cada 500) y no afecta al SNC. Afectación neurológica variable según tipo. Forma juvenil asociada a deterioro neurológico de inicio tardío.")
43
ENFERMEDAD DE GAUCHER Déficit de beta-glucocerebrósido-hidroxilasa
Esplenomegalia = primer signo clínico Hiperesplenismo e insuf. de la médula ósea Pancitopenia: anemia, leucopenia y trombocitopenia Hepatomegalia moderada Alteraciones radiológicas: huesos largos con deformidad de Erlenmeyer

45
ENFERMEDAD DE GAUCHER

46
ENFERMEDAD DE GAUCHER: Alteraciones Radiológicas

47
ENFERMEDAD DE GAUCHER DIAGNÓSTICO
Clínico: esplenomegalia y anemia leve no explicada Aspiración de médula ósea Genética molecular : cromosoma1

48
E. GAUCHER

49
ENFERMEDAD DE GAUCHER: Médula Ósea

50
ENFERMEDAD DE GAUCHER TRATAMIENTO - Esplenectomía en desuso
Reposición enzimática: Ceredase ó cerezyme U/kg x 4 semanas i.v. Y luego la mitad cada 2 semanas

51
Lecodistrofia Metacromática
LEUCODISTROFIA METACROMÁTICA Deficit de arilsulfatasa A = Depósito de sulfatos en los lisosomas de la sustancia blanca FORMA INFANTIL - Forma grave y frecuente Irritabilidad, incapacidad para andar, hiperextension de las rodillas. Convulsiones mioclónicas. Atrofia óptica, tetraparesia espástica FORMA JUVENIL Ataxia, deterioro y dificultades emocionales Curso lento Aparece a los 20 años FORMA DEL ADULTO. Tardía

52
LEUCODISTROFIA METACROMÁTICA
Los sulfatos no degradados se depositan en la sustancia blanca = desmielinización Reducción en el número de células de oligodendroglia No hay afectación visceral ni de la médula ósea

53
LEUCODISTROFIA METACROMÁTICA

54
LEUCODISTROFIA METACROMÁTICA
DIAGNÓSTICO - Clínica Biopsia del nervio safeno externo con depósitos metacromáticos Orina con excreción de sulfatos EMG:disminución de la conducción nerviosa TAC y RM: atenuación de la sustancia blanca Estudios enzimáticos de los leucocitos y cultivos de fibroblastos cutáneos Genética molecular: cromosoma 22

56
LEUCODISTROFIA METACROMÁTICA
TRATAMIENTO No existe El transplante de médula ósea mejora los niveles enzimáticos,pero no el deterioro neurológico PRONÓSTICO - En la forma infantil tardía viven 2-4 años y en la juvenil 4-6 años después del diagnóstico

57
HASTA otro dia

Presentaciones similares





>")


>")














 ¿ENFERMEDADES RARAS?>")