Descargar la presentación
La descarga está en progreso. Por favor, espere

1
HEMOFILIA ENFERMEDAD DE VON WILLEBRAND

2
PREVALENCIA Hemofilia A 1/ Hemofilia B 1/ VWD 3% población

3
Propéptido Subunidad Madura
protómero dominios funcionales ligandos Factor von Willebrand Propéptido Subunidad Madura I S D D2 D’ D3 A1A2A3 D4BC1C2 S I D’ D A1A2A3 D4BC1C2 S I RGD Colágeno GPIIb-IIIa FVIII GPIb Heparina Heparina Colágeno Sulfátidos

4
vWF Ensamble Multímeros
RETICULO ENDOPLASMICO GOLGI vWF Ensamble Multímeros Furina Sadler JE, 2005

5
vWF Ensamble y Catabolismo
Proteolisis aumentada (vWD2A) ADAMTS-13 kproteolisis Inicial Plasma vWF Ensamble y Catabolismo ksecreción kdepuración Depuración aumentada (vWD1 Vicenza) Sadler JE, 2005
 ADAMTS-13. kproteolisis. Inicial. Plasma. vWF Ensamble y Catabolismo. ksecreción. kdepuración. Depuración aumentada (vWD1 Vicenza) Sadler JE,")
6
FACTOR VIII A1 A2 B A3 C1 C2 Secreción Preactivación Activación
NH 2 A1 A2 B A3 C1 C2 Secreción Preactivación Activación Inactivación 200kD kD IIa 1648 90kD HRG kD IIa 740 IIa 50kD kD kD 372 APC IIa Xa Xa 1689 45kD kD 336 1721 Lollar P & Parker CG, 1989

7
FVIII-VWF FVIIIa FVIII VWF N N C C S-S S-S IIa, FXa Vlot et al., 1998
A A2 FVIIIa C C A3 IIa, FXa 372 A2 FVIII 740 A1 C1 A3 B VWF C2 1689 N N C C S-S S-S Vlot et al., 1998

8
FVIII/VWF - Propiedades
Adhesión Endotelio/Sub Transporta FVIII Clínica Hemofilia vWD Producción S. Hepáticos C. Endoteliales Megacariocitos Gen Cromosoma X Cromosoma 12 Molécula Heterodímero Multímero Función Coagulación Procofactor

9
FVIII/VWF - Nomenclatura
Factor VIII Antígeno FVIII:Ag Función FVIII Factor von Willebrand Proteína Madura VWF VWF:Ag Cofactor Ristocetina VWF:RCo Unión al Colágeno VWF:CB Unión al Factor VIII VWF:FVIIIB ISTH

10
HEMOFILIA A Recesiva Ligada al sexo (gen FVIIICr X) PORTADORA
HEMOFILICO

11
HEMOFILIA Modo de Herencia

12
HEMOFILIA Modo de Herencia

14
HEMOFILIA Diagnóstico “sencillo”
- APTT prolongado - FVIII disminuido

15
HEMOFILIA - Clasificación
Severa < 1% VIII Moderada 1-5% VIII Leve >5% VIII

16
HEMOFILIA Diagnóstico de Portadoras

17
PORTADORA OBLIGADA Padre hemofílico Hijo hemofílico y antecedentes
(vía materna) 2 o más hijos, sin antecedentes familiares No requiere estudio
 2 o más hijos, sin antecedentes familiares. No requiere estudio.")
18
PORTADORA POTENCIAL Hijo hemofílico, sin antecedentes familiares
Sin hijos y con antecedentes (vía materna) Dificultad diagnóstica
 Dificultad diagnóstica.")
19
PORTADORAS Lyonización
Ovulo Fertilizado Heterocigota Clones XM XP XP XM XM XP XP XM XP XM XM XP XM XP

20
PORTADORAS HEMOFILIA Factor VIII Portadoras Normales probabilidad
FVIII

21
Bioensayos DESVENTAJAS Probabilidad Error por lyonización extrema

22
Análisis de ADN VENTAJAS Certeza Independiente de lyonización

23
Análisis de ADN Alteración heterogénea Afecta diferentes regiones
Complejo Alteración heterogénea Afecta diferentes regiones del gen No aplicable a todos los casos

24
IDENTIFICACION INDIRECTA
No Certeza Análisis de segregación de RFLP ? -/ /- PORTADORA (-) asociado al defecto
 asociado al defecto.")
25
50% hemofílicos severos
IDENTIFIACION DIRECTA Certeza Inversión del intrón 22 ↓ 50% hemofílicos severos

26
IDENTIFIACION DIRECTA
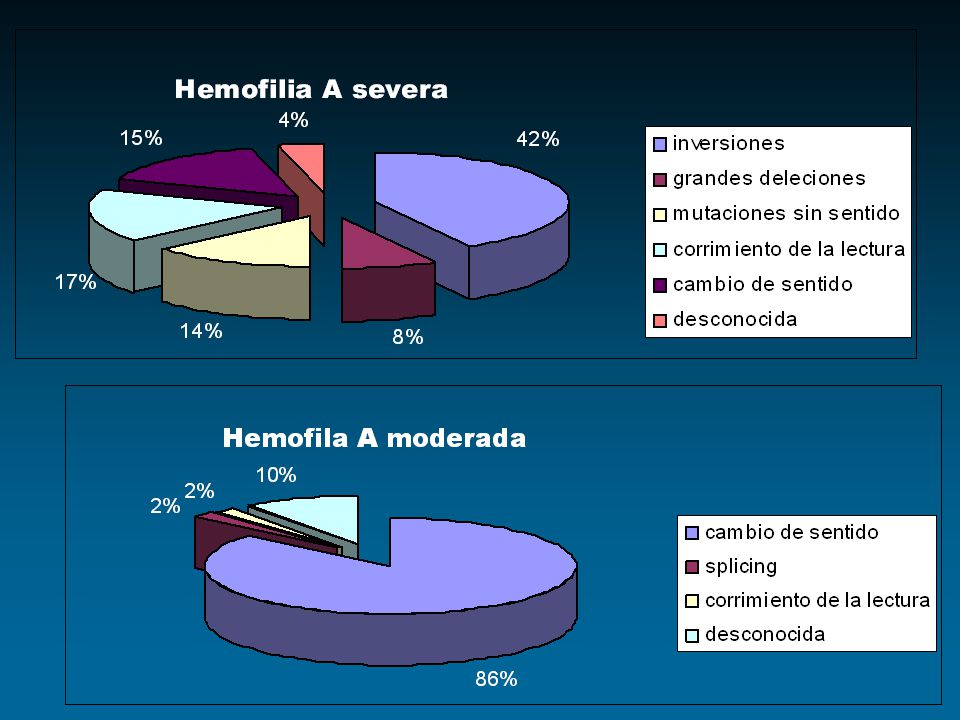
Inversiones Deleciones Inserciones Mutaciones “sin sentido” Mutaciones “sentido falso”

27
HEMOFILIA B Recesiva Ligada al sexo (gen FIXCr X) PORTADORA
HEMOFILICO

28
HEMOFILIA - Clasificación
Severa < 1% IX Moderada 1-5% IX Leve >5% IX

29
HEMOFILIA B Diagnóstico “sencillo”
- APTT prolongado - FIX disminuido

30
Propéptido Subunidad Madura
protómero dominios funcionales ligandos Factor von Willebrand Propéptido Subunidad Madura I S D D2 D’ D3 A1A2A3 D4BC1C2 S I D’ D A1A2A3 D4BC1C2 S I RGD Colágeno GPIIb-IIIa FVIII GPIb Heparina Heparina Colágeno Sulfátidos

31
VWD Clasificación Revisada Sadler E, 1994
Tipo 1 Deficiencia cuantitativa parcial Tipo 2 Alteración cualitativa Tipo 3 “Ausencia”de VWF

32
VWF Cromosoma 12 (12p1212pter)
VWD Genética VWF Cromosoma (12p1212pter)
")
33
VWD – TIPOS Frecuencia (%)
Tuddenham (134) 75 19 6 Lenk (111) 76 12 Nilson (106f) 70 10 20 Hoyer (116) 71 23 Awidi (65) 59 29,5 11,5 Berliner (60) 62 9 29 Castaman G, 2003
 Lenk (111) Nilson (106f) Hoyer (116) Awidi (65) ,5. 11,5. Berliner (60) Castaman G,")
34
VWD Serie 1885 Pacientes FVIII 61,6% VWF:Ag 62,2% TS 58,4%
(Tipo I 91% ) FVIII 61,6% VWF:Ag 62,2% TS 58,4% Woods AI, 2001
 FVIII 61,6% VWF:Ag 62,2% TS 58,4% Woods AI,")
35
Modificadires Ambientales Enfermedad primariamutación
VWF Expresión Variable Penetrancia Incompleta Chance Modificadires Ambientales edad, género, trauma Enfermedad primariamutación ? Genes modificadores Niveles Variables VWF Difícil Diagnóstico Ginsburg D, 2001

36
VWF-Variabilidad Individual Familiar

37
VWF-Variabilidad Factores 40% no genéticos 60% genéticos

38
VWF-Variabilidad Factores no-genéticos Edad Estrés Ejercicio
H. Tiroideas Estrógenos F.Aguda

39
VWF-Variabilidad Factores genéticos Grupo ABO 20-30% variabilidad
Grupo “O” VWF Hipótesis Glicosilación Alteración procesamiento estabilidad secreción

40
VWD Distribución según grupo ABO “AB” 0,6% difícil diagnóstico
“O” 70% más sintomático > frecuencia de consulta Woods AI, 2001

41
VWF Pruebas Específicas
Cuantitativa Cualitativa VWF:Ag SI NO VWF:RCo SI SI VWF:CB SI SI Multímeros SEMI SI

42
VWF-ELISA 1 2 3 4 1. Anti-VWF inmovilizado
a-vWF vWF a-vWF-HRP Color-Abs 2. Incubación con muestra o control 1. Anti-VWF inmovilizado 3. Incubación con anti-VWF-conjugado 4. Incubación con sustrato cromogénico

43
VWF:RCo Plaquetas normales Plasma del paciente

44
Agregación c/Ristocetina
VWF:RCo Plaquetas normales Plasma del paciente Ristocetina Agregometría Agregación c/Ristocetina

45
Agregación c/Ristocetina
VWF:RCo Plaquetas normales Plasma del paciente Ristocetina Agregometría Agregación c/Ristocetina PRP Plaquetas del paciente Plasma del paciente

46
Agregación c/Ristocetina
VWF:RCo Plaquetas normales Plasma del paciente Ristocetina Agregometría Agregación c/Ristocetina PRP Plaquetas del paciente Plasma Ristocetina Agregometría

47
Agregación c/Ristocetina
Normal Agregación c/Ristocetina vWD-Tipo 1 vWD-Tipo 2A vWD-Tipo 3 Kasper C, 2005

48
VWF:CB Unión Colágeno 1 2 3 4 1. Inmovilización 5 6 2. Bloqueo
Colágeno Tipo III BSA 3 a-vWFconejo Muestra 4 Esferas poliestireno IgG F(ab’)2* 1. Inmovilización 5 6 2. Bloqueo 3. Colágeno VWF Citometría de flujo 4. VWF a-VWF 5. a-VWF IgGF(ab’)2fluoresceína 6. Adquisición
2* 1. Inmovilización Bloqueo. 3. Colágeno VWF. Citometría. de flujo. 4. VWF a-VWF. 5. a-VWF IgGF(ab’)2fluoresceína. 6. Adquisición.")
49
Actividad VWF-ELISA a-vWF vWF a-vWF-HRP Color-Abs 1. AcMo a-vWFepitope funcional inmovilizado 2. Incubación con muestra o control 3. Incubación con anti-vWF-conjugado 4. Incubación con sustrato cromogénico

50
Agregación c/Ristocetina
Kasper C, 2005

51
VWF-Análisis Multimérico
grandes intermedios-pequeños (banda 1) (banda 2) Línea Línea Línea 3 Lane 3> 2B Normal 2A
 (banda 2) Línea 1 Línea 2 Línea 3. Lane 3> 2B Normal 2A.")
52
vWD-Tipo 2 Clasificación Revisada Sadler E, 1994
2A función plaquetaria Multímeros APM 2M función plaquetaria c/Multímeros APM 2N afinidad VWF-FVIII* *símil Hemofilia A, autosómica 2B afinidad VWF-GPIb-IX

53
Propéptido Subunidad Madura
protómero dominios funcionales ligandos Factor von Willebrand Propéptido Subunidad Madura I S D D2 D’ D3 A1A2A3 D4BC1C2 S I D’ D A1A2A3 D4BC1C2 S I X RGD Colágeno GPIIb-IIIa FVIII GPIb Heparina Heparina Colágeno Sulfátidos

54
VWF:FVIIIB Enlace FVIII VWF
a-vWF vWF FVIII Color-Abs CaCl2 0,4M 1. Anticuerpo a-vWF inmovilizado 2. Incubación con muestra o control 3. Incubación FVIII purificado/CaCl2 4. Incubación con sustrato cromogénico

55
VWF:FVIIIB Enlace FVIII VWF
-3.50 -3.25 -3.00 -2.75 -2.50 -2.25 -2.00 0.50 0.55 0.60 0.65 0.70 0.75 0.80 0.85 0.90 0.95 Referencia Paciente 1 Paciente 2 Absorbancia 405nm

56
2N-Variante Normandy Biología Molecular. -. Secuenciación. -. Enz
2N-Variante Normandy Biología Molecular - Secuenciación - Enz. restricción

57
VWF- Arg19Trp (C2594T) Exón 18 Enzima BspMI Mutación Normal
1-244 pb pb pb Normal pb 96 pb Mutado pb 1 2 3 Mutación Normal

58
Screening VWD Estrategia Diagnóstica * T. Sangría APTT (fVIII)
Adhesividad Tiempo de obstrucción (PFA) *no permiten excluir VWD
 *no permiten excluir VWD.")
59
Diagnóstico VWD Estrategia Diagnóstica -VWF plasma/plaquetas
VWF:Ag VWF:RCo VWF:CB -FVIII en plasma

60
Clasificación VWD Estrategia Diagnóstica Multímeros RIPA
Unión GP plaquetarias VWF:FVIIIB Biología Molecular

61
vWD- Importancia Dignóstica
VWF:RCo Prueba más sensible VWF:Ag CorrelacionaVWF:RCo Ratio=1Tipo1 Ratio<1Tipo2 FVIII Util para el manejo clínico T. Sangría CorrelacionaVWF:RCo (T1) Detecta defecto plaquetario RIPA Detecta Tipo 2B Castaman G, 2003
 Detecta defecto plaquetario. RIPA. Detecta Tipo 2B. Castaman G,")
62
vWD- Importancia Dignóstica
A.Multimérico DiscriminaTipo 2M, 2A, 2B VWFplaquetario Predice respuesta DDAVPTipo 1 VWF:FVIIIB DetectaTipo 2N VWF:CB CorrelacionaVWF:RCo (T1) Sensibilidad colágeno CT PFA-100 Especificidad (?) Castaman G, 2003
 Sensibilidad colágeno. CT PFA-100. Especificidad ( ) Castaman G,")
63
HEMOFILIA VON WILLEBRAND Diagnóstico Diferencial

64
Enfermedad de von Willebrand
Cofactor de ristocetina * Antígeno de factor vW o N Factor VIII o N Tiempo de Sangría o N *excepto la variante 2N

65
Hemofilia Cofactor de ristocetina N Antígeno de factor vW N
Factor VIII Tiempo de Sangría N

66
Hemofilia-VWD 2N Hemofilia VWD2N VWF:RCo N N Factor VIII
VWF:FVIIIB N

67
VWD Laboratorio Diagnóstico Control de terapéutica

68
COAGULPATIAS Orientación Diagnóstica

69
PRUEBAS V. INTRINSECA TTPA V. EXTRINSECA TP V. FINAL TT FC XII XIIa
XI XIa Ca2+ V. EXTRINSECA TP X Xa Va FL Ca2+ FT VIIa Ca2+ IX IXa VIIIa FL Ca2+ Fibrinógeno M. Fibrina Fibrina V. FINAL TT II IIa XIIIa Activación

70
INTERPRETACION Pruebas DEFECTO (probable) ↑TP, APTT, TT ↑APTT, TS TP
APTT, TT, TS Normales V. Extrínseca (FVII) Déficit de factores Vit K Dep. Hepatopatía TP, APTT TT, TS Normales V. Común, Déficit de factores Vit K Dep., Hepatopatía TP, APTT, TT, TS Hepatopatía, CID ↑TP, APTT, TT TS Normal V. Final Fibrinógeno APTT TP, TT, TS Normal V. Intrínseca (FVIII, FIX, FXI, FXII) Hemofilia, VWD ↑APTT, TS TP, TT Normales VWD
 Déficit de factores Vit K Dep. Hepatopatía. TP, APTT. TT, TS Normales. V. Común, Déficit de factores Vit K Dep., Hepatopatía. TP, APTT, TT, TS. Hepatopatía, CID. ↑TP, APTT, TT. TS Normal. V. Final. Fibrinógeno. APTT. TP, TT, TS Normal. V. Intrínseca (FVIII, FIX, FXI, FXII) Hemofilia, VWD. ↑APTT, TS. TP, TT Normales. VWD.")
71
INTERPRETACION Pruebas DEFECTO (probable) ↑TP, APTT, TT ↑APTT, TS TP
APTT, TT, TS Normales V. Extrínseca (FVII) Déficit de factores Vit K Dep. Hepatopatía TP, APTT TT, TS Normales V. Común, Déficit de factores Vit K Dep., Hepatopatía TP, APTT, TT, TS Hepatopatía, CID ↑TP, APTT, TT TS Normal V. Final Fibrinógeno APTT TP, TT, TS Normal V. Intrínseca (FVIII, FIX, FXI, FXII) Hemofilia, VWD ↑APTT, TS TP, TT Normales VWD
 Déficit de factores Vit K Dep. Hepatopatía. TP, APTT. TT, TS Normales. V. Común, Déficit de factores Vit K Dep., Hepatopatía. TP, APTT, TT, TS. Hepatopatía, CID. ↑TP, APTT, TT. TS Normal. V. Final. Fibrinógeno. APTT. TP, TT, TS Normal. V. Intrínseca (FVIII, FIX, FXI, FXII) Hemofilia, VWD. ↑APTT, TS. TP, TT Normales. VWD.")
72
INTERPRETACION Pruebas DEFECTO (probable) ↑TP, APTT, TT ↑APTT, TS TP
APTT, TT, TS Normales V. Extrínseca (FVII) Déficit de factores Vit K Dep. Hepatopatía TP, APTT TT, TS Normales V. Común, Déficit de factores Vit K Dep., Hepatopatía TP, APTT, TT, TS Hepatopatía, CID ↑TP, APTT, TT TS Normal V. Final Fibrinógeno APTT TP, TT, TS Normal V. Intrínseca (FVIII, FIX, FXI, FXII) Hemofilia, VWD ↑APTT, TS TP, TT Normales VWD
 Déficit de factores Vit K Dep. Hepatopatía. TP, APTT. TT, TS Normales. V. Común, Déficit de factores Vit K Dep., Hepatopatía. TP, APTT, TT, TS. Hepatopatía, CID. ↑TP, APTT, TT. TS Normal. V. Final. Fibrinógeno. APTT. TP, TT, TS Normal. V. Intrínseca (FVIII, FIX, FXI, FXII) Hemofilia, VWD. ↑APTT, TS. TP, TT Normales. VWD.")
73
INTERPRETACION Pruebas DEFECTO (probable) ↑TP, APTT, TT ↑APTT, TS TP
APTT, TT, TS Normales V. Extrínseca (FVII) Déficit de factores Vit K Dep. Hepatopatía TP, APTT TT, TS Normales V. Común, Déficit de factores Vit K Dep., Hepatopatía TP, APTT, TT, TS Hepatopatía, CID ↑TP, APTT, TT TS Normal V. Final Fibrinógeno APTT TP, TT, TS Normal V. Intrínseca (FVIII, FIX, FXI, FXII) Hemofilia, VWD ↑APTT, TS TP, TT Normales VWD
 Déficit de factores Vit K Dep. Hepatopatía. TP, APTT. TT, TS Normales. V. Común, Déficit de factores Vit K Dep., Hepatopatía. TP, APTT, TT, TS. Hepatopatía, CID. ↑TP, APTT, TT. TS Normal. V. Final. Fibrinógeno. APTT. TP, TT, TS Normal. V. Intrínseca (FVIII, FIX, FXI, FXII) Hemofilia, VWD. ↑APTT, TS. TP, TT Normales. VWD.")
74
INTERPRETACION Pruebas DEFECTO (probable) ↑TP, APTT, TT ↑APTT, TS TP
APTT, TT, TS Normales V. Extrínseca (FVII) Déficit de factores Vit K Dep. Hepatopatía TP, APTT TT, TS Normales V. Común, Déficit de factores Vit K Dep., Hepatopatía TP, APTT, TT, TS Hepatopatía, CID ↑TP, APTT, TT TS Normal V. Final Fibrinógeno APTT TP, TT, TS Normal V. Intrínseca (FVIII, FIX, FXI, FXII) Hemofilia, VWD ↑APTT, TS TP, TT Normales VWD
 Déficit de factores Vit K Dep. Hepatopatía. TP, APTT. TT, TS Normales. V. Común, Déficit de factores Vit K Dep., Hepatopatía. TP, APTT, TT, TS. Hepatopatía, CID. ↑TP, APTT, TT. TS Normal. V. Final. Fibrinógeno. APTT. TP, TT, TS Normal. V. Intrínseca (FVIII, FIX, FXI, FXII) Hemofilia, VWD. ↑APTT, TS. TP, TT Normales. VWD.")
75
INTERPRETACION Pruebas DEFECTO (probable) ↑TP, APTT, TT ↑APTT, TS TP
APTT, TT, TS Normales V. Extrínseca (FVII) Déficit de factores Vit K Dep. Hepatopatía TP, APTT TT, TS Normales V. Común, Déficit de factores Vit K Dep., Hepatopatía TP, APTT, TT, TS Hepatopatía, CID ↑TP, APTT, TT TS Normal V. Final Fibrinógeno APTT TP, TT, TS Normal V. Intrínseca (FVIII, FIX, FXI, FXII) Hemofilia, VWD ↑APTT, TS TP, TT Normales VWD
 Déficit de factores Vit K Dep. Hepatopatía. TP, APTT. TT, TS Normales. V. Común, Déficit de factores Vit K Dep., Hepatopatía. TP, APTT, TT, TS. Hepatopatía, CID. ↑TP, APTT, TT. TS Normal. V. Final. Fibrinógeno. APTT. TP, TT, TS Normal. V. Intrínseca (FVIII, FIX, FXI, FXII) Hemofilia, VWD. ↑APTT, TS. TP, TT Normales. VWD.")
76
INTERPRETACION Pruebas DEFECTO (probable) ↑TP, APTT, TT ↑APTT, TS TP
APTT, TT, TS Normales V. Extrínseca (FVII) Déficit de factores Vit K Dep. Hepatopatía TP, APTT TT, TS Normales V. Común, Déficit de factores Vit K Dep., Hepatopatía TP, APTT, TT, TS Hepatopatía, CID ↑TP, APTT, TT TS Normal V. Final Fibrinógeno APTT TP, TT, TS Normal V. Intrínseca (FVIII, FIX, FXI, FXII) Hemofilia, VWD ↑APTT, TS TP, TT Normales VWD
 Déficit de factores Vit K Dep. Hepatopatía. TP, APTT. TT, TS Normales. V. Común, Déficit de factores Vit K Dep., Hepatopatía. TP, APTT, TT, TS. Hepatopatía, CID. ↑TP, APTT, TT. TS Normal. V. Final. Fibrinógeno. APTT. TP, TT, TS Normal. V. Intrínseca (FVIII, FIX, FXI, FXII) Hemofilia, VWD. ↑APTT, TS. TP, TT Normales. VWD.")
77
Gracias

Presentaciones similares