Descargar la presentación
La descarga está en progreso. Por favor, espere

1
TEMA 7. INMUNODEFICIENCIAS ASOCIADAS A DEFECTOS EN LOS LINFOCITOS T

2
Inmunodeficiencias primarias de las células T: por defectos heredable en la diferenciación y función de las células T: Células T Precursores Timo Células presentadoras Clínica: Generalmente manifestaciones en el primer año de vida Mayor susceptibilidad a infecciones: microorganismos intracelulares Es frecuente un retraso en el crecimiento Hay generalmente un incremento en la frecuencia de alergias, autoinmunidad y linfomas

3
Estudio de laboratorio: Sospecha clínica:
Retraso del crecimiento, infecciones recurrentes por patógenos oportunistas como Candida albicans, Pneumocystis jiroveci o CMV a edades muy tempranas Diarreas crónicas, infecciones bacterianas recurrentes que afectan a múltiples sitio o persistentes a pesar del tto Diagnóstico: Inmunofenotipado por citometría de flujo Estudios funcionales in vitro de las células T

4
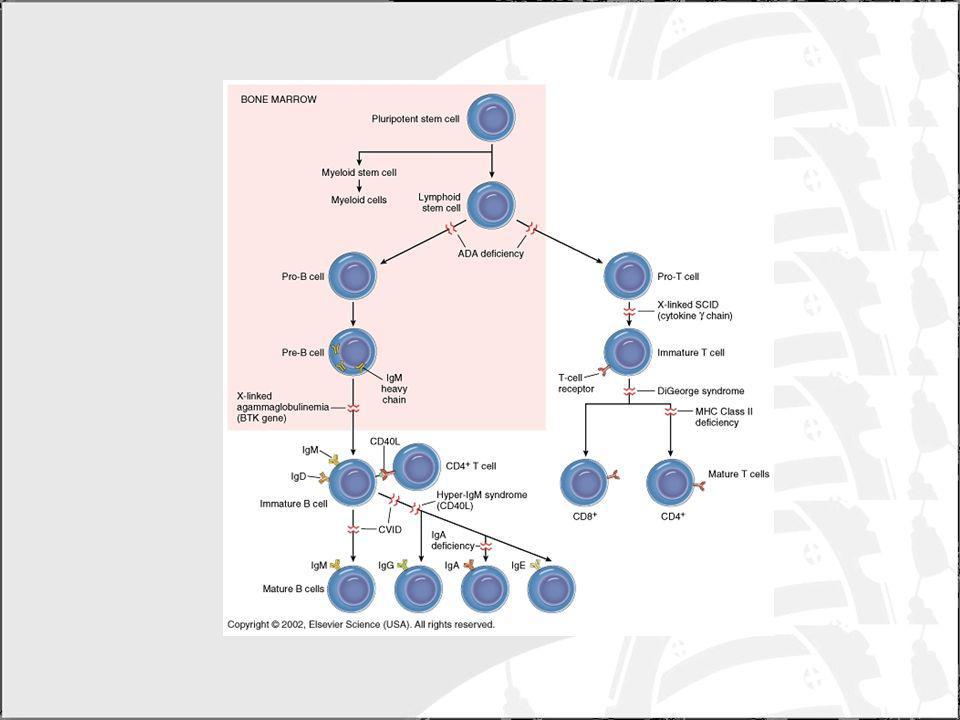
Inmunodeficiencias combinadas severas (SCID)
Clasificación: Inmunodeficiencias combinadas severas (SCID) Defectos en la recombinación VDJ Deficiencia den la adenosina desaminasa (ADA) Defecto en la señalización del TCR Inmunodeficiencias con células T detectables pero que son deficientes (inmunodeficiencias combinadas: CID) Inmunodeficiencias de las células T con defectos no inmunes asociados Síndrome de DiGeorge Síndrome de Wiskott-Aldrich (WAS)
 Defectos en la recombinación VDJ. Deficiencia den la adenosina desaminasa (ADA) Defecto en la señalización del TCR. Inmunodeficiencias con células T detectables pero que son deficientes (inmunodeficiencias combinadas: CID) Inmunodeficiencias de las células T con defectos no inmunes asociados. Síndrome de DiGeorge. Síndrome de Wiskott-Aldrich (WAS)")
6
Inmunodeficiencias combinadas severas (SCID)
Se producen por defectos en los linfocitos T que se acompañan o no de una diferenciación anormal de linfocitos B y células NK. Producen una muerte temprana si no hay tto con TMO. Frecuencia: 1: a 1: nacimientos vivos. Aparición temprana de infecciones respiratorias e intestinales. Son frecuentes los organismos oportunistas: Pneumocystis jiroveci y Aspergillus, organismos intracelulares: Listeria y Legionella, virus: herpesvirus y adenovirus. Llevan al diagnóstico: candidiasis orales, diarreas persistentes y retraso en el crecimiento. No diferencias clínicas excepto en el momento en que aparecen las infecciones.

7
Inmunodeficiencias combinadas severas (SCID)
Manifestaciones clínicas no infecciosas: GVHD Se puede producir porque el paciente no puede eliminar células alogénicas Fuente de células alogénicas: Linfocitos T de origen materno Transfusiones El 50% de los pacientes con SCID presentan células T de origen materno en cantidades variables. Fenotipo normal Son poco respondedores a mitógenos Generalmente producen síntomas leves o asintomáticos Problemas que conllevan las células maternas: Dificultan el diagnóstico Pueden obstaculizar el TMO en donantes HLA no idénticos Muy peligroso la realización de transfusiones: síndrome GVGD agudo mortal

8
Inmunodeficiencias combinadas severas (SCID)
Otras manifestaciones clínicas: Hipoplasia de los órganos linfoides secundarios Timo hipotrófico: ausencia de componentes linfoides y deficiente diferenciación de las células epiteliales. No hay corpúsculos de Hassal Otras características: Número muy bajo de células T maduras. Si hay células B no son funcionales. Si no son tratados con TMO mueren a los 1-2 años de vida Diagnóstico: Con antecedentes familiares o esporádicos Infecciones severas o de organismos raros Ausencia de ganglios linfáticos detectables Linfocitopenia Ensayos de proliferación y ensayos enzimáticos (ADA…) Confirmación genética si es posible
 Confirmación genética si es posible.")
9
Inmunodeficiencias combinadas severas (SCID)
Manejo del paciente: Realizar diagnóstico inmunológico de forma rápida. No deben recibir vacunas con microorganismos vivos. Transfusiones: irradiar, deplecionar linf T, descartar CMV. Aislar al paciente. Profilaxis con antibiótico. El TMO es el único tto curativo.

10
Inmunodeficiencias combinadas severas (SCID)
Clasificación: Defectos en la recombinación VDJ SCID autosómico recesivo Señalización defectiva de los receptores de citoquinas SCID ligada al cromosoma X Deficiencia en JAK3 Deficiencia en el IL-7R Deficiencia en la adenosina desaminasa (ADA) Defecto en la señalización del TCR
 Defecto en la señalización del TCR.")
11
Defectos en la recombinación VDJ
SCID autosómico recesivo (T-, B-, NK+): Este fenotipo se encuentra en el 20% de los pacientes con SCID Se distinguen dos grupos: Defectos en Artemis (células radiosensibles) Defectos en RAG-1/RAG-2. Los genes RAG-1/RAG-2 son necesarios para realizar la recombinación entre los segmentos VDJ Artemis participa en procesos de recombinación y es necesaria para la recombinación de los segmentos VDJ y en procesos de reparación de DNA Síndrome de Omenn: se produce por mutaciones hipomórficas de estos genes
: Este fenotipo se encuentra en el 20% de los pacientes con SCID. Se distinguen dos grupos: Defectos en Artemis (células radiosensibles) Defectos en RAG-1/RAG-2. Los genes RAG-1/RAG-2 son necesarios para realizar la recombinación entre los segmentos VDJ. Artemis participa en procesos de recombinación y es necesaria para la recombinación de los segmentos VDJ y en procesos de reparación de DNA. Síndrome de Omenn: se produce por mutaciones hipomórficas de estos genes.")
13
Señalización defectiva de los receptores de citoquinas
SCID ligada al cromosoma X (T-, B+, NK-) Este fenotipo se encuentra en el 50% de los pacientes con SCID. Tienen linfocitos B pero son anómalos fenotípica y funcionalmente requieren tto con inmunoglobulinas. Las células NK no están bien diferenciadas. Se produce por un defecto en la cadena del IL-2R SCID por déficit en el JAK3 (T-, B+, NK-) Mismo fenotipo que la anterior pero con herencia autosómica recesiva Participa en la transducción de la señal del IL-2R
 Este fenotipo se encuentra en el 50% de los pacientes con SCID. Tienen linfocitos B pero son anómalos fenotípica y funcionalmente requieren tto con inmunoglobulinas. Las células NK no están bien diferenciadas. Se produce por un defecto en la cadena del IL-2R. SCID por déficit en el JAK3 (T-, B+, NK-) Mismo fenotipo que la anterior pero con herencia autosómica recesiva. Participa en la transducción de la señal del IL-2R.")
14
Señalización defectiva de los receptores de citoquinas
SCID por deficiencia en el IL-7R (T-, B+, NK+): Hay una ausencia selectiva de linfocitos T maduros. La maduración de las células B y NK no está afectada La presencia de esta cadena es necesaria para el desarrollo de los linfocitos T
: Hay una ausencia selectiva de linfocitos T maduros. La maduración de las células B y NK no está afectada. La presencia de esta cadena es necesaria para el desarrollo de los linfocitos T.")
15
Deficiencia en la adenosina desaminasa (ADA)
Tienen un fenotipo (T-, B-, NK-) Constituye el 20% de las SCID En ausencia de ADA se acumula hasta mil veces más de dATP en las células linfoides El dATP inhibe a la ribonucleótido reductasa. Los linfocitos son más sensibles debido a que apenas tienen actividad 5’nucleotidasa Gen 12 exones
 Constituye el 20% de las SCID. En ausencia de ADA se acumula hasta mil veces más de dATP en las células linfoides. El dATP inhibe a la ribonucleótido reductasa. Los linfocitos son más sensibles debido a que apenas tienen actividad 5’nucleotidasa. Gen 12 exones.")
16
TERAPIA GENICA

17
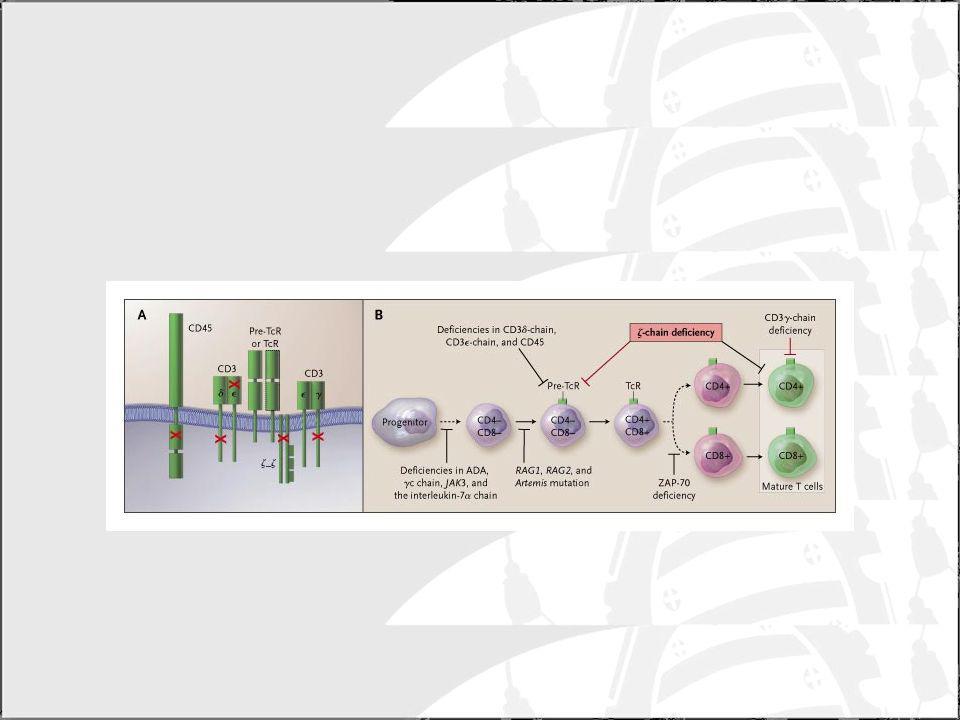
Defectos en la señalización del TCR
Deficiencias en el CD3 y Deficiencias en el CD45

18
Inmunodeficiencias combinadas (CID)
Los pacientes presentan células T detectables pero que no son funcionales. No suelen presentar en los primeros años de vida infecciones que pongan en peligro su vida. Presentan, además de infecciones, otras complicaciones como autoinmunidad, alergia y cáncer que están causadas por la inmunodeficiencia. Las infecciones no suelen ser la primera manifestación clínica. Las infecciones suelen ser del tracto respiratorio (producen bronquiectasias), diarreas, infecciones del SNC (bacterianas o víricas) y candidiasis cutánea o de mucosas. 2/3 desarrollan infecciones virales repetidas y/o severas, especialmente del grupo de los herpesvirus. Mas de la mitad tienen algún tipo de proceso autoinmune. Al menos el 50% algún tipo de alergia (IgE elevada). Esperanza de vida 5-10 años de media.
, diarreas, infecciones del SNC (bacterianas o víricas) y candidiasis cutánea o de mucosas. 2/3 desarrollan infecciones virales repetidas y/o severas, especialmente del grupo de los herpesvirus. Mas de la mitad tienen algún tipo de proceso autoinmune. Al menos el 50% algún tipo de alergia (IgE elevada). Esperanza de vida 5-10 años de media.")
19
Inmunodeficiencias combinadas (CID)
Diagnóstico: Presentan infecciones de repetición asociadas con procesos autoinmunes y alergia En los ensayos funcionales las células T tienen una pobre función Los niveles de inmunoglobulinas pueden estar disminuidos o ser normales pero las respuesta humoral suele ser poco eficiente frente a las infecciones

20
Inmunodeficiencias combinadas (CID)
Clasificación: Deficiencias en las moléculas HLA de clase II Deficiencia en TAP-1 y TAP2 Deficiencia en CD3 y Deficiencias en la activación de las células T Deficiencia en ZAP-70 Síndrome de Ommen Deficiencia en la purina nucleósido fosforilas (PNP)
")
21
Inmunodeficiencias combinadas (CID)
Deficiencia en las moléculas HLA de clase II: Es poco frecuente y se da en el área mediterránea Los síntomas aparecen el primer año de vida: Infecciones y/o enfermedades autoinmunes Fallecen a los 4 años de media, aunque pueden llegar a la edad adulta con poca sintomatología Diagnóstico: ausencia de moléculas HLA-DR, -DQ, y –DP en las superficie de todas las células que expresan constitutivamente moléculas HLA de clase II (células B, monocitos y DCs) o después de la activación por el IFN (células T, fibroblastos) Se han descrito varios defectos en factores de transcripción asociados a esta enfermedad
 o después de la activación por el IFN (células T, fibroblastos) Se han descrito varios defectos en factores de transcripción asociados a esta enfermedad.")
22
Inmunodeficiencias combinadas (CID)
Deficiencia en las moléculas HLA de clase II (contin.): Factores de transcripción asociados: RFXAP RFX5 RFXANK CIITA
: Factores de transcripción asociados: RFXAP. RFX5. RFXANK. CIITA.")
23
Inmunodeficiencias combinadas (CID)
Deficiencia en TAP-1 y TAP-2: Tienen poca expresión de moléculas de clase I y linfopenia de células T CD8. Está afectado el complejo TAP (transporta péptidos desde el citoplasma al RE para ser cargados en las moléculas de clase I) Diagnóstico: se detecta una reducción en veces la expresión de moléculas de clase I en las células mononucleares del la sangre. Las células CD8 que quedan son activas frente a virus. Suelen tener incrementado el nivel de inmunoglobulinas que además funcionan adecuadamente.
 Diagnóstico: se detecta una reducción en veces la expresión de moléculas de clase I en las células mononucleares del la sangre. Las células CD8 que quedan son activas frente a virus. Suelen tener incrementado el nivel de inmunoglobulinas que además funcionan adecuadamente.")
24
Inmunodeficiencias combinadas (CID)
Deficiencia en CD3 y z: Son muy poco frecuentes. Los niveled de CD3 en los linfocitos el de un 1-50% de normal El número de células T es normal aunque no funcionan adecuadamente (proliferación disminuida) Fenotipo variable incluso dentro de la misma familia Deficiencias en la activación de las células T: Deficiencia en ZAP-70 Produce deficiencia en señalización a través del TCR Número bajo de CD8 y normal de CD4 (aunque no responden a la activación). Nivel bajo de inmunoglobulinas y deficiencia humoral
 Fenotipo variable incluso dentro de la misma familia. Deficiencias en la activación de las células T: Deficiencia en ZAP-70. Produce deficiencia en señalización a través del TCR. Número bajo de CD8 y normal de CD4 (aunque no responden a la activación). Nivel bajo de inmunoglobulinas y deficiencia humoral.")
25
Inmunodeficiencias combinadas (CID)
Síndrome de Ommen: Comienzo temprano tras el nacimiento con eritroderma difuso y alopecia en cabeza y cejas Retraso en el crecimiento con infecciones que ponen en peligro la vida Linfocitosis (fenotipo Th2) e hipereosinofilia Piel e intestino infiltrados por linfocitos T Ganglios linfátitos agrandados pero desprovistos de linfocitos (tienen macrófagos) Apenas inmunoglobulinas, excepto IgE que está aumentada Fenotipo T-, B- (típico de las SCID) Se produce por mutaciones hipomórficas parciales de RAG1/RAG2 o del gen de Artemis
 e hipereosinofilia. Piel e intestino infiltrados por linfocitos T. Ganglios linfátitos agrandados pero desprovistos de linfocitos (tienen macrófagos) Apenas inmunoglobulinas, excepto IgE que está aumentada. Fenotipo T-, B- (típico de las SCID) Se produce por mutaciones hipomórficas parciales de RAG1/RAG2 o del gen de Artemis.")
26
Inmunodeficiencias combinadas (CID)
Deficiencia en la purina nucleósido fosforilasa (PNP): Es muy infrecuente (50 casos) Agresiva inmunodeficiencia celular. 1/3 autoinmunidad. 2/3 manifestaciones neurológicas Se produce inhibición de la ribonucleótido reductasa (por acúmulo de dGTP)
: Es muy infrecuente (50 casos) Agresiva inmunodeficiencia celular. 1/3 autoinmunidad. 2/3 manifestaciones neurológicas. Se produce inhibición de la ribonucleótido reductasa (por acúmulo de dGTP)")
27
Inmunodeficiencias de células T asociadas a defectos no inmunes
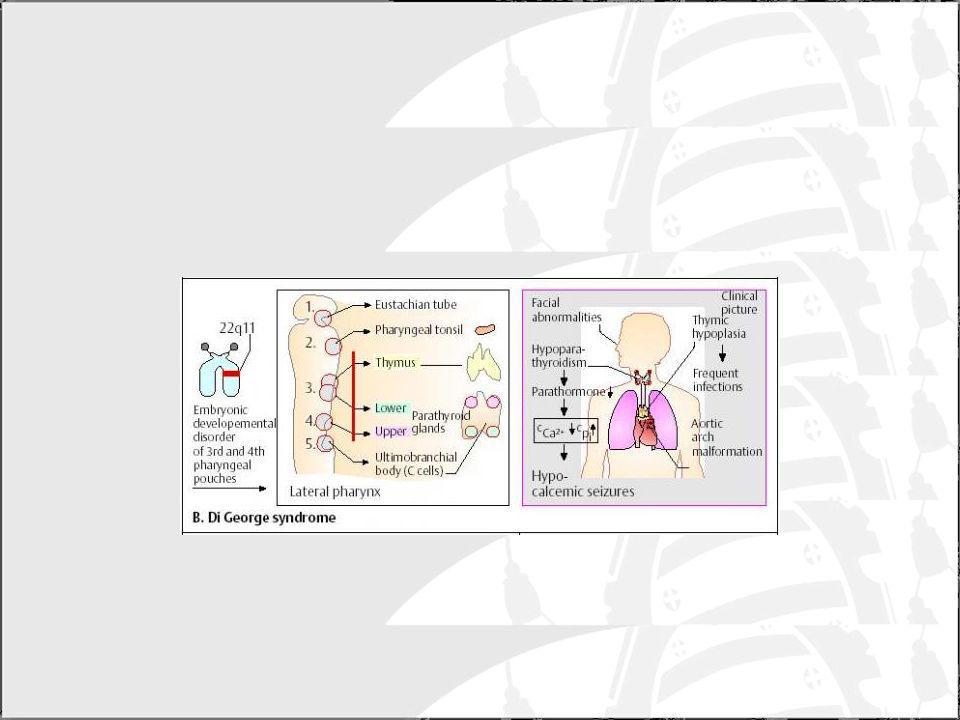
Síndrome de DiGeorge Defecto en el desarrollo de las bolsas faríngeas 3ª y 4ª. Tienen defectos tímicos, paratiroideos y cardiovasculares. Aspecto facial característico: micrognacia, orejas pequeñas, hipertelorismo ocular. Es común el retraso mental. Clínica variable: Aplasia completa y número células T muy bajo. Aplasia parcial (más frecuente) e inmunodeficiencia leve. Se ha encontrado una zona de susceptibilidad en el cromosoma 22 de 250 kb que incluye genes candidatos. Las formas más graves pueden requerir TMO.
 e inmunodeficiencia leve. Se ha encontrado una zona de susceptibilidad en el cromosoma 22 de 250 kb que incluye genes candidatos. Las formas más graves pueden requerir TMO.")
29
Inmunodeficiencias de células T asociadas a defectos no inmunes
Síndrome de Wiskott-Aldrich (WAS) Ligada al cromosoma X. Frecuencia 1: nacimientos vivos Se caracteriza por inmunodeficiencia asociada con trombocitopenia, plaquetas anormales y eccema. Tienden a sangrar incluso en los primeros años de vida. Producción de anticuerpos deficiente: infección por microorganismos encapsulados. Deficiencia en las células T con reducción gradual del número de células T (pueden producirse infecciones oportunistas) Manifestaciones autoinmunes sobre todo de autoanticuerpos frente a las plaquetas y anemia hemolítica. Riesgo elevado de tumores, sobre todo linfomas.
 Ligada al cromosoma X. Frecuencia 1: nacimientos vivos. Se caracteriza por inmunodeficiencia asociada con trombocitopenia, plaquetas anormales y eccema. Tienden a sangrar incluso en los primeros años de vida. Producción de anticuerpos deficiente: infección por microorganismos encapsulados. Deficiencia en las células T con reducción gradual del número de células T (pueden producirse infecciones oportunistas) Manifestaciones autoinmunes sobre todo de autoanticuerpos frente a las plaquetas y anemia hemolítica. Riesgo elevado de tumores, sobre todo linfomas.")
30
Inmunodeficiencias de células T asociadas a defectos no inmunes
Síndrome de Wiskott-Aldrich (WAS) (Contin.) El TMO con donantes idénticos posibilidad de curación en más del 90% de los casos. Se debe a defecto en el gen de la proteína WAS (WASP) que está implicada en la reorganización de la actina del citoesqueleto de los leucocitos y megacariocitos.
 (Contin.) El TMO con donantes idénticos posibilidad de curación en más del 90% de los casos. Se debe a defecto en el gen de la proteína WAS (WASP) que está implicada en la reorganización de la actina del citoesqueleto de los leucocitos y megacariocitos.")
Presentaciones similares














 ESPECÍFICAS (Respuesta inmunitaria) – La unión antígeno anticuerpo es específica.>")




