Descargar la presentación
La descarga está en progreso. Por favor, espere

1
Mónica Romero Riquelme
CITODIAGNÓSTICO EN SÍNDROMES MIELODISPLÁSICOS Mónica Romero Riquelme 5 de febrero de 2009

2
Introducción Los SMD son una enfermedad clonal heterogénea Características: displasia, citopenias, hemato-poyesis ineficaz, alto riesgo de evolución a leucemia aguda Edad media al diagnóstico : 70 años Variada Importancia fundamental del citodiagnóstico

3
Concepto de SMD

4
Concepto de CRSI

5
Indices pronósticos

6
Indices pronósticos

7
Indices pronósticos

8
Indices pronósticos

9
Criterios de respuesta

10
Criterios de respuesta

11
Valoración de la displasia
Fundamental para el diagnóstico Evaluar en cada línea celular Evaluar en SP y MO ( cuantitativa y cualitativa) Calidad de la muestra: frotis sin anticoagulante. Si no es posible:EDTA realizar extensiones en máximo 2 horas
 Calidad de la muestra: frotis sin anticoagulante. Si no es posible:EDTA. realizar extensiones en máximo 2 horas.")
12
Diseritropoyesis Valorar 100 eritroblastos Mayor o = 10% eritroblastos dismórficos: displasia eritroide OMS Sangre periférica: anisocitosis; punteado basófilo; cuerpos Howell-Jolly; anillos Cabot

13
Diseritropoyesis Médula ósea:
Alteraciones nucleares:; multinuclearidad; picnosis; puentes internucleares; irregularidad del contorno nuclear; hiperlobulación nuclear

14
Diseritropoyesis Médula ósea:
Alteraciones citoplasmáticas: vacuolización citoplasmática; puentes intercitoplasmáticos; punteado basófilo; disrelación núcleo/citoplasma; distribución irregular de la hemoglobina PAS positividad de eritroblastos Tinción de Perls: sideroblastos anillados > o= 15% Alteraciones de mayor grado: puentes internuclares; multinuclearidad; PAS +; sideroblastos anillados

15
Disgranulopoyesis Valorar 200 células ( mejor SP)
Mayor o = 10% granulocitos dismórficos: displasia granulocítica OMS Alteraciones nucleares: bisegmentación (pseudo Pelger-Huet); hiposegmentación nuclear; hipersegmentación nuclear; clumping cromatínico; apéndices nucleares
; hiposegmentación nuclear; hipersegmentación nuclear; clumping cromatínico; apéndices nucleares.")
16
Gold standard: granulocitos hipolobulados agranulares
Disgranulopoyesis Alteraciones citoplasmáticas: hipogranulación; asincronismo madurativo; granulación tóxica; granulación pseudo-Chediak; cuerpos de Dohle ; bastones de Auer Gold standard: granulocitos hipolobulados agranulares

17
Distrombopoyesis Mayor 0 = 10% megacariocitos dismórficos: displasia megacariocítica OMS Sangre periférica: macroplaquetas (pseudo Bernard Soulier); plaquetas hipogranuladas ( plaquetas grises); plaquetas agranuladas ( plaquetas azules)
; plaquetas hipogranuladas ( plaquetas grises); plaquetas agranuladas ( plaquetas azules)")
18
Distrombopoyesis Valorar 30 megacariocitos
Médula ósea: megacariocitos monolobulados; asincronismo madurativo; núcleos dispersos; micromegacariocitos mononucleados (núcleo excéntrico y redondo) Alteraciones de mayor grado: micromegacariocitos mononucleados, megacariocitos hipolobulados; agrupación de micromegacariocitos
 Alteraciones de mayor grado: micromegacariocitos mononucleados, megacariocitos hipolobulados; agrupación de micromegacariocitos.")
19
Anemia refractaria con sideroblastos anillados
Clasificación FAB 1982 Clasificación OMS 2001 Clasificación OMS 2008 Anemia refractaria Anemia refractaria con sideroblastos anillados Anemia refractaria con exceso de blastos Anemia refractaria con exceso de blastos en transformación Leucemia mielomonocítica crónica Anemia refractaria Anemia refractaria con sideroblastos anillados Citopenia(s) refractaria(s) con displasia multilínea Citopenia refractaria con diaplasia multilínea y sideroblastos anillados Anemia refractaria con exceso de blastos tipo I Anemia refractaria con exceso de blastos tipo II Sindrome mielodisplásico sociado a del (5q) como única alteración genética Sindrome mielodisplásico no clasificable Citopenia refractaria con displasia unilínea: anemia refractaria; neutropenia refractaria; trombopenia refractaria Anemia refractaria con sideroblastos anillados Citopenia refractaria con displasia multilínea Anemia refractaria con exceso de blastos tipo 1 Anemia refractaria con exceso de blastos tipo 2 Sindrome mielodisplásico asociado a del (5q) aislada Sindrome mielodisplásico no clasisficable
 refractaria(s) con displasia multilínea. Citopenia refractaria con diaplasia multilínea y sideroblastos anillados. Anemia refractaria con exceso de blastos tipo I. Anemia refractaria con exceso de blastos tipo II. Sindrome mielodisplásico sociado a del (5q) como única alteración genética. Sindrome mielodisplásico no clasificable. Citopenia refractaria con displasia unilínea: anemia refractaria; neutropenia refractaria; trombopenia refractaria. Anemia refractaria con sideroblastos anillados. Citopenia refractaria con displasia multilínea. Anemia refractaria con exceso de blastos tipo 1. Anemia refractaria con exceso de blastos tipo 2. Sindrome mielodisplásico asociado a del (5q) aislada. Sindrome mielodisplásico no clasisficable.")
20
Novedades OMS 2008 Nueva categoría: citopenia refractaria con displasia unilínea: AR, NR y TR Definición de sideroblastos anillados: 5 o+ gránulos, ubicación perinuclear Mejor definición de SMSI

21
Citopenia refractaria con displasia unilínea - anemia refractaria
Sangre periférica Médula ósea Genética Inmunofe notipo Otros 1.S.eritroide: Anemia N-N o N-M, Hb <10 Anisopoiquilo-citosis grado variable 2.S.granulocítica:N 3.S.plaquetar: N 4.Monocitos < 1x109/L 5.Blastos <1% 1.Hipercelular 2.S.eritroide: aumentada; displasia > o = 10%, sideroblastos anillados <15% 3.S.mieloide: N 4.S.megaca: N 5.Blastos <5% 6.No bastones Auer 1. 25% casos alteraciones 2. Más frecuentes: +8; alt 5; alt 7, del (20q) No patrón definido % SMD 2. Media sobrevida: 66 meses 3. Evol a LA: 5% / 2 años
 No patrón definido % SMD. 2. Media sobrevida: 66 meses. 3. Evol a LA: 5% / 2 años.")
22
Citopenia refractaria con displasia unilínea – neutropenia refractaria
Recuento neutrófilos : < 1800 cels/ mm 3 Displasia granulocítica > o= 10% Blastos en SP < 1% Blastos en MO < 5% Citopenia refractaria con displasia unilínea – trombopenia refractaria Recuento plaquetar: < / mm3 Displasia megacariocítica > o = 10% Blastos en SP < 1% Blastos en MO < 5%

23
AR con sideroblastos anillados
Sangre periférica Médula ósea Genética Inmunofe notipo Otros 1. S.eritroide: anemia N-N o N-M, Hb<10 gr/dL 2. S.granulocítica:N 3. S.megacariocí: N 4. Blastos <1% 5. Monocitos<1x109 1. Hipercelular 2. S.eritroide: aumentada, con displasia > o = 10%, sideroblastos anillados > o = 15%, PAS – 3. Blastos < 5% 4. S.mieloide: N 5. S.megacario: N 6. No bastones de Auer 1. Alteraciones 5-20% casos No patrón definido % SMD 2. Mayor frecuencia en varones 3. Edad media: años 4. Evol a LA: 2% 5. Sobrevida: meses

24
Citopenia refractaria con displasia multilínea
Sangre periférica Médula ósea Genética Inmunofe notipo Otros 1. Citopenia(s): Hb <10 gr/dL RAN < 1800 cels / mm3 Plaquetas < /mm3 2. Displasia > o = 10% en 2 o + líneas mieloides 3. Blastos <1% 4. Monocitos <1x109/L 1. MO Hipercelular 3. Sideroblastos anillados > o < 15% 4. Blastos < 5% 5. No bastones de Auer 1. Alteraciones 50% casos 2. Alt más frecuentes: +8, alt 7, del (5q) ; del (20q) no patrón definido 1. 30% SMD 2.Sideroblas tos anillados no alteran pronóstico 3. Media edad: 70 años 4. > frecuen varones 5. Media sobrevida: 30 meses 6. Evol a LA: 10% / 2años
: Hb <10 gr/dL. RAN < 1800 cels / mm3. Plaquetas < /mm3. 2. Displasia > o = 10% en 2 o + líneas mieloides. 3. Blastos <1% 4. Monocitos <1x109/L. 1. MO Hipercelular. 3. Sideroblastos anillados > o < 15% 4. Blastos < 5% 5. No bastones de Auer. 1. Alteraciones 50% casos. 2. Alt más frecuentes: +8, alt 7, del (5q) ; del (20q) no patrón definido % SMD. 2.Sideroblas tos anillados no alteran pronóstico. 3. Media edad: 70 años. 4. > frecuen varones. 5. Media sobrevida: 30 meses. 6. Evol a LA: 10% / 2años.")
25
AREB TIPO 1 Y 2 Sangre periférica Médula ósea Genética Inmunofe-
notipo Otros 1.Displasia uni o multilínea 2.s.granulocítica en todos los estadios madurativos 3. No hiato leucémico 4. AREB 1: >1 y < 5% de blastos 5. AREB 2: 5-19% blastos 1. MO hipercelular 2. s.eritroide: displasia (> rasgos megalob); < 15% de sideroblastos anillados 3. S.megacariocítica: predominio de megac pequeños e hipolobulados 4. AREB 1: 5-9% blastos 5. AREB 2: 10-19% blastos; presencia de bastones Auer; blastos en MO < 5% pero 2-4% en SP 30-50% casos con alteraciones genéticas 2. Alt más frecuentes: +8; -5; 5q-; -7; 7q-; 20q-: cariotipo complejo 1. Aumento de células CD34+ y/o CD 117+ Otros +: CD38; HLA-DR; CD13; CD33 2. 20% expres aberrante de CD7 y 10% CD56 3. Micromegac: + para CD61 y CD42b 1. 40% de SMD 2. Edad media 50 años 3. Evol a LA: AREB 1: 25% ; AREB 2 : 33% 4. Sob media: AREB 1 : 16 meses; AREB 2: 9 meses
; < 15% de sideroblastos anillados. 3. S.megacariocítica: predominio de megac pequeños e hipolobulados. 4. AREB 1: 5-9% blastos. 5. AREB 2: 10-19% blastos; presencia de bastones Auer; blastos en MO < 5% pero 2-4% en SP % casos con alteraciones genéticas. 2. Alt más frecuentes: +8; -5; 5q-; -7; 7q-; 20q-: cariotipo complejo. 1. Aumento de células CD34+ y/o CD 117+ Otros +: CD38; HLA-DR; CD13; CD % expres aberrante de CD7 y 10% CD Micromegac: + para CD61 y CD42b % de SMD. 2. Edad media. 50 años. 3. Evol a LA: AREB 1: 25% ; AREB 2 : 33% 4. Sob media: AREB 1 : 16 meses; AREB 2: 9 meses.")
26
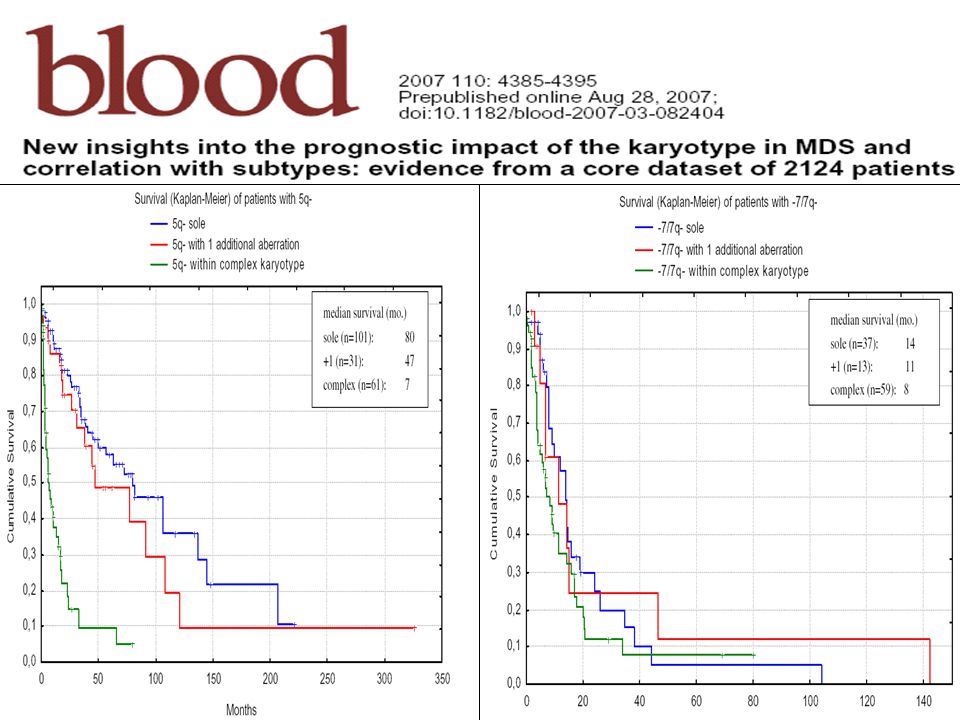
Síndrome 5q- Sangre periférica Médula ósea Genética Inmunofe notipo
Otros 1.S eritroide: anemia(> grave); N-M 2.S granulocítica: N o disminuida, sin rasgos de displasia 3. S plaquetar: N o trombocitosis ½ casos 4. Blastos < 1% 1. MO hiper o normocelular 2. S eritroide: hipoplasia; displasia rara 3. S mieloide: Displasia rara 4.megacariocítica: Aumentada; megacariocitos de tamaño pequeño con núcleo único o hipolobulado ( micromega) 5.Frecuentes agregados linfoides 6.Blastos < 5% 7.No bastones Auer 1.del(5q) como alteración única. 2. Pérdida de un gen supresor EGR1y CTNNA1 3. Bandas más afectadas: q31-33 4. alteración genética agregada : no Sd 5q- ( excepto –Y) 5. Mutación JAK2 V617F en algunos casos No patrón definido 1. Mayor frecuencia en mujeres 2. Import dep- transf (80% dg) 3. Edad media 67 años 4. Sob global media 145 meses 5. Evol a LA < 10% 6. Resp a Lenalidom
; N-M. 2.S granulocítica: N o disminuida, sin rasgos de displasia. 3. S plaquetar: N o trombocitosis ½ casos. 4. Blastos < 1% 1. MO hiper o normocelular. 2. S eritroide: hipoplasia; displasia rara. 3. S mieloide: Displasia rara. 4.megacariocítica: Aumentada; megacariocitos de tamaño pequeño. con núcleo único o hipolobulado. ( micromega) 5.Frecuentes agregados linfoides. 6.Blastos < 5% 7.No bastones Auer. 1.del(5q) como alteración única. 2. Pérdida de un gen supresor EGR1y CTNNA1. 3. Bandas más afectadas: q alteración genética agregada : no Sd 5q- ( excepto –Y) 5. Mutación JAK2 V617F en algunos casos. No patrón definido. 1. Mayor frecuencia. en mujeres. 2. Import dep- transf. (80% dg) 3. Edad media 67 años. 4. Sob global media 145 meses. 5. Evol a LA < 10% 6. Resp a Lenalidom.")
27
SMD no clasificable 2001 Concepto mal definido 2008
Incluía neutropenia o trombopenia refractaria No % de displasia 2008 Sangre periférica: citopenia(s); blastos < o = 1% Médula ósea : MO hipercelular; displasia < 10% en 1 ó más líneas mieloides y una citogenética alterada presunta de SMD; blastos MO <5%; displasia unilínea con pancitopenia; pacientes con CRDU o CRDM pero 1% de blastos en SP Incidencia desconocida, pronóstico no establecido
; blastos < o = 1% Médula ósea : MO hipercelular; displasia < 10% en 1 ó más líneas mieloides y una citogenética alterada presunta de SMD; blastos MO <5%; displasia unilínea con pancitopenia; pacientes con CRDU o CRDM pero 1% de blastos en SP. Incidencia desconocida, pronóstico no establecido.")
28
SMD-formas especiales
SMD hipocelular MO hipocelular desde el diagnóstico, gral AR o AREB 5-10% de los SMD de novo, > relacionados a tratamiento Mayor frecuencia en mujeres, > evolución a LMA Diagnóstico diferencial más importante: anemia aplásica Importancia de la biopsia de MO y tipaje de blastos con CD34

29
SMD CON FIBROSIS O HIPERFIBRÓTICO
SMD-formas especiales SMD CON FIBROSIS O HIPERFIBRÓTICO 10-20% SMD de novo, > SMD relacionados a tratamiento Sangre periférica: citopenia(s), escasos dacriocitos y leucoeritorblastosis escasa MO: Fibrosis principalmente reticulínica; displasia de al menos 2 líneas mieloides;MO hipercelular, con importante proliferación de megacariocitos Diagnóstico diferencial: SAMPC; LMA-M7; panmielosis aguda con fibrosis. Importancia CD34; CD117; MPO Citogenética: alteraciones frecuentes y cariotipo complejo Pronóstico desfavorable
, escasos dacriocitos y leucoeritorblastosis escasa. MO: Fibrosis principalmente reticulínica; displasia de al menos 2 líneas mieloides;MO hipercelular, con importante proliferación de megacariocitos. Diagnóstico diferencial: SAMPC; LMA-M7; panmielosis aguda con fibrosis. Importancia CD34; CD117; MPO. Citogenética: alteraciones frecuentes y cariotipo complejo. Pronóstico desfavorable.")
30
SMD CON CORRELACIÓN CLÍNICO/CITOGENÉTICA
SMD-formas especiales SMD CON CORRELACIÓN CLÍNICO/CITOGENÉTICA 1. Anomalías del cromosoma 17: (17p-) o i(17)(q20). Alteración p53 1-5% de los SMD. Características de SMD y SMPC Disgranulopoyesis: hipolobulación núcleo; abundantes y pequeñas vacuolas citoplasmáticas; abundancia de monocitos y macrófagos; aumento de blastos Elevado riesgo de transformación a leucemia aguda 2. Anomalías del cromosoma 3: inv(3)(q21q26); t(3,5)(q25;q33); t(3,3)(q21;q26). Alteración EVI1 2% de los SMD,> relacionados a tratamiento Aumento de blastos; plaquetas N o aumentadas; dismegacariopoyesis ( micromegacariocitos)
 o i(17)(q20). Alteración p % de los SMD. Características de SMD y SMPC. Disgranulopoyesis: hipolobulación núcleo; abundantes y pequeñas vacuolas citoplasmáticas; abundancia de monocitos y macrófagos; aumento de blastos. Elevado riesgo de transformación a leucemia aguda. 2. Anomalías del cromosoma 3: inv(3)(q21q26); t(3,5)(q25;q33); t(3,3)(q21;q26). Alteración EVI1. 2% de los SMD,> relacionados a tratamiento. Aumento de blastos; plaquetas N o aumentadas; dismegacariopoyesis ( micromegacariocitos)")
31
SMD CON CORRELACIÓN CLÍNICO/CITOGENÉTICA
SMD-formas especiales SMD CON CORRELACIÓN CLÍNICO/CITOGENÉTICA 3. Anomalías del cromosoma 7: -7 y 7q- Dismegariocitopoyesis ( micromegacariocitos muy dismórficos) Infecciones graves Evolución desfavorable SMD con eosinofilia 7% de los SMD Mayor frecuencia en hombres Aumento de blastos Eosinófilos atípicos ( formas nucleares irregulares; granulación pseudobasófila; vacuolización citoplasmática ) > o = 5% en MO
 Infecciones graves. Evolución desfavorable. SMD con eosinofilia. 7% de los SMD. Mayor frecuencia en hombres. Aumento de blastos. Eosinófilos atípicos ( formas nucleares irregulares; granulación pseudobasófila; vacuolización citoplasmática ) > o = 5% en MO.")
32
SMD secundario Pronóstico desfavorable
50% con MO hipocelular y/o fibrótica al diagnóstico Acentuada inmadurez de las líneas hematopoyéticas Secundaria a agentes alquilantes: Aparición a los 5 – 10 años del tratamiento, con displasia multilínea Leucemización más frecuente a LMA M1-M2 FAB Anomalías genéticas de cromosomas 7 y 5 Resistencia total a terapia Secundaria a inhidores de topoisomerasa II Aparición 2 –3 años del tratamiento Leucemización más frecuente a LMA M4-M5 Alteraciones genéticas balanceadas 11q23, 21q22 Respuesta parcial a quimioterapia

33
SMD infantil Muy infrecuente, < 5% neoplasias hematológicas en < 14 años Mayor frecuencia de MO hipocelular, menor frecuencia de SMD bajo riesgo OMS 2008 Citopenia refractaria de la infancia: citopenia persistente; blastos en MO < 5% y en SP < 2%, displasia en al menos 10% de la serie comprometida o rasgos displásicos en 2 o más líneas mieloides. Infrecuentes sideroblastos anillados. 75% MO hipocelular, gran valor diagnóstico de la biopsia ( clusters de precursores eritroides inmaduros, micromegacariocitos) Monosomía 7: alteración genética más frecuente
 Monosomía 7: alteración genética más frecuente.")
34
Morfología en SMD

35
Morfología en SMD

37
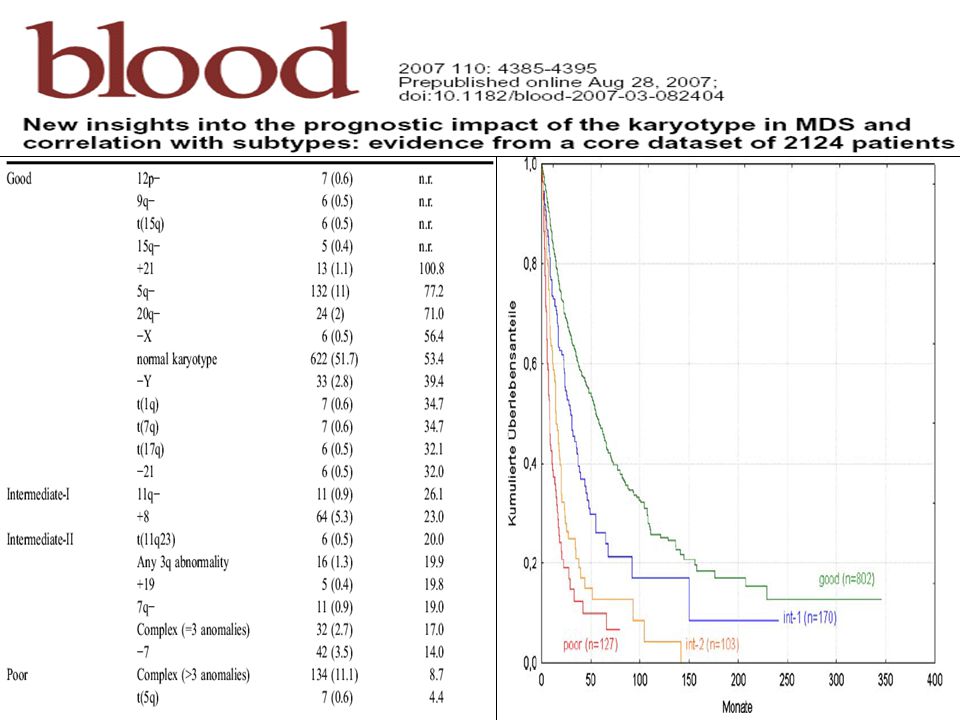
Citogenética en SMD 50% alteraciones genéticas en SMD
Perfil genética característico: alteraciones no balanceadas (pérdida de material genético) inactivación de genes supresores Importancia pronóstico (IPSS): 3 grupos Riesgo favorable: cariotipo normal; ; del(5q)-Y; del(20q) Riesgo intermedio: +8, < 3 alteraciones cromosómicas Riesgo desfavorable: cariotipo complejo; 7- Cariotipo normal: 50% Alteraciones más frecuentes: Del(5q) aislada: banda más afectada q31, frecuencia 16-28% Alteraciones cromosoma 7: % Trisomía 8: 8-10%
 inactivación de genes supresores. Importancia pronóstico (IPSS): 3 grupos. Riesgo favorable: cariotipo normal; ; del(5q)-Y; del(20q) Riesgo intermedio: +8, < 3 alteraciones cromosómicas. Riesgo desfavorable: cariotipo complejo; 7- Cariotipo normal: 50% Alteraciones más frecuentes: Del(5q) aislada: banda más afectada q31, frecuencia 16-28% Alteraciones cromosoma 7: 11-26% Trisomía 8: 8-10%")
38
Tipo y condiciones de la muestra: elección MO
Tipo y condiciones de la muestra: elección MO. SP si blastos 10-20%; tubo heparina y volumen 1-3 mL; tº ambiente no más de 24 horas; cultivos cortos ( hrs) Cariotipo-Metafases a analizar: 20; ISCN2005 FISH: no reemplaza cariotipo. Indicación imprescindible: al diagnóstico, escasas o nulas metafases o cromosomas de pobre calidad. Tejido de elección: MO Núcleos a examinar: 200 Sondas imprescindibles: 5q31; cen7; 7q31, cen8 Sondas opcionales: 20q; p53(17p13); cromosoma Y
 Cariotipo-Metafases a analizar: 20; ISCN2005. FISH: no reemplaza cariotipo. Indicación imprescindible: al diagnóstico, escasas o nulas metafases o cromosomas de pobre calidad. Tejido de elección: MO. Núcleos a examinar: 200. Sondas imprescindibles: 5q31; cen7; 7q31, cen8. Sondas opcionales: 20q; p53(17p13); cromosoma Y.")
39
Citogenética en SMD

42
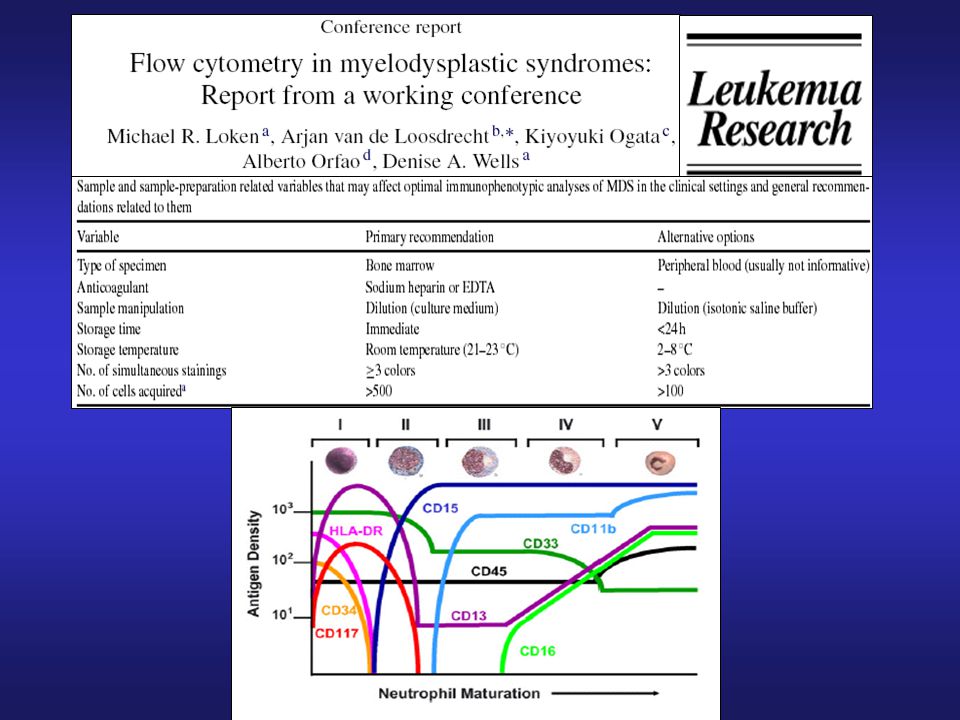
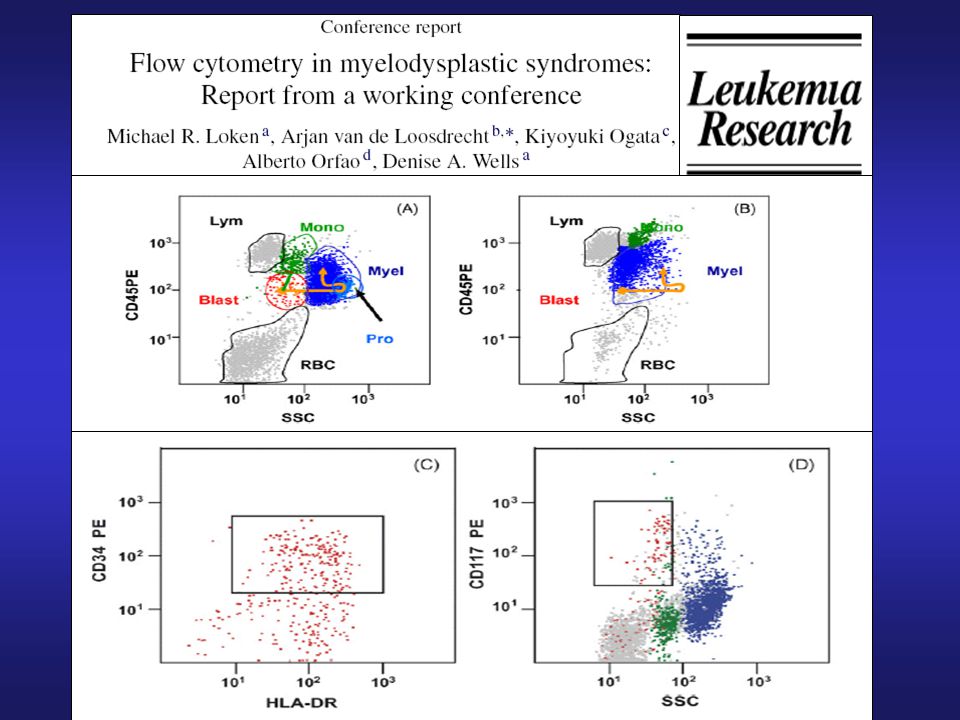
Inmunofenotipo en SMD

46
Sindromes mielodisplásicos/mieloproliferativos
Leucemia mielomonocítica crónica Leucemia mieloide crónica atípica, BCR/ABL negativa Leucemia mielomonocítica juvenil Sindrome mielodisplásico/mieloproliferativo no clasificable

47
Leucemia mielomonocítica crónica
Monocitosis persistente en SP: > 1x109/L Ausencia de cromosoma Ph o gen de fusión BCR/ABL o reordenamiento PDGFRA o PDGFRB < 20% blastos (mieloblastos,monoblastos,promonocitos) en SP y MO Displasia en una o más líneas mieloides Si displasia ausente o mínima: uno de los siguientes: alteración clonal citogenética o molecular adquirida, monocitosis persistente por al menos 3 meses, excluir causas de monocitosis Subtipos: LMMC-1: blastos <5% SP ; <10% MO LMMC-2: blastos 5-19% SP,10-19% MO o bastones Auer + con < 20% blastos LMMC-eosinofilia: eosinófilos >=1,5x109/L
 en SP y MO. Displasia en una o más líneas mieloides. Si displasia ausente o mínima: uno de los siguientes: alteración clonal citogenética o molecular adquirida, monocitosis persistente por al menos 3 meses, excluir causas de monocitosis. Subtipos: LMMC-1: blastos <5% SP ; <10% MO. LMMC-2: blastos 5-19% SP,10-19% MO o bastones Auer + con < 20% blastos. LMMC-eosinofilia: eosinófilos >=1,5x109/L.")
48
Leucemia mieloide crónica atípica BCR/ABL neg
Leucocitosis en SP > o = 13 x109/L, debido a aumento de neutrófilos y sus precursores, con prominente disgranulopoyesis Ausencia de cromosoma Ph o gen de fusión BCR/ABL o de reordenamiento PDGFRA o PDGFRB Precursores neutrófilos (promielocitos,mielocitos,metamielocitos) > o = 10% de los leucocitos Basófilos < 2% de los leucocitos Monocitos <10% de los leucocitos MO hipercelular, con proliferación y displasia granulocítica, con o sin displasia en serie eritroide y megacariocítica Blastos en SP y MO < 20%
 > o = 10% de los leucocitos. Basófilos < 2% de los leucocitos. Monocitos <10% de los leucocitos. MO hipercelular, con proliferación y displasia granulocítica, con o sin displasia en serie eritroide y megacariocítica. Blastos en SP y MO < 20%")
49
Leucemia mielomonocítica juvenil
Monocitosis SP > 1x109/L Blastos (incluídos promonocitos) < 20% en SP y MO Ausencia de cromosoma Ph o del gen de fusión BCR/ABL Dos o más de los siguientes: Hemoglobina F aumentada para la edad Granulocitos inmaduros en SP Leucocitos > 10x109/L Alteración cromosómica clonal ( por ej monosomía7) Hipersensibilidad a GM-CSF de progenitores mieloides in vitro
 < 20% en SP y MO. Ausencia de cromosoma Ph o del gen de fusión BCR/ABL. Dos o más de los siguientes: Hemoglobina F aumentada para la edad. Granulocitos inmaduros en SP. Leucocitos > 10x109/L. Alteración cromosómica clonal ( por ej monosomía7) Hipersensibilidad a GM-CSF de progenitores mieloides in vitro.")
50
Sindrome mielodisplásico/mieloproliferativo
no clasificable Presencia de datos clínicos, morfológicos y de laboratorio de una de las categorías de SMD con <20% de blastos en SP y MO + rasgos mieloproliferativos prominentes + ausencia de SMD o SMPC previo, ausencia de tratamiento reciente con citostáticos o factores de crecimiento. Ausencia de cromosoma Ph o del gen de fusión BCR/ABL o del reordenamiento PDGFRA o PDGFRB o FGFR1, ausencia de del(5q), t(3,3)(q21;q26) o inv(3)(q21q26) o el paciente tiene rasgos de SMD y SMPC que no puede ser asignado a ninguna categoría de éstos (RARS-T)
, t(3,3)(q21;q26) o inv(3)(q21q26) o el paciente tiene rasgos de SMD y SMPC que no puede ser asignado a ninguna categoría de éstos (RARS-T)")
51
Mónica Romero Riquelme
CITODIAGNÓSTICO EN SÍNDROMES MIELODISPLÁSICOS Mónica Romero Riquelme 5 de febrero de 2009

Presentaciones similares