Descargar la presentación
La descarga está en progreso. Por favor, espere

1
Dr. Roberto Carrillo Briceño Médico internista hematólogo
BALANZA HEMOSTÁTICA Dr. Roberto Carrillo Briceño Médico internista hematólogo

2
FISIOLOGÍA MEDULA ÓSEA SANGRE PERIFÉRICA ÓRGANOS BALANZA HEMOSTÁTICA

3
BALANZA HEMOSTÁTICA FASE VASCULAR FASE PLAQUETARIA FASE PLASMÁTICA

4
HEMOSTASIA NORMAL Depende de un complejo sistema.
Interacción del endotelio vascular con las proteínas del plasma y las plaquetas. Las enfermedades adquiridas o heredita-rias de este sistema pueden generar una hemorragia o trombosis anormales.

5
HEMOSTASIA PRIMARIA Es una respuesta inmediata, segundos o minutos, pero inestable a la agresión. Clave: plaquetas y vasos sanguíneos. Plaquetas – moléculas de la matriz m.b. una molecula de enlace, el factor de von Willebrand.

6
Hemostasia secundaria Coagulación
Es una respuesta lenta, minutos u horas, pero produce un coágulo definitivo de fibrina. Repara el vaso sanguíneo. La lesión hística inicia el proceso ( el factor VII queda expuesto al factor hístico )
")
7
Mecanismos Hemostáticos
1. Hemostasia 1ª Adherencia y agregación plaquetaria al endotelio. 2. Hemostasia 2ª Formación del coágulo de fibrina

8
Reguladores naturales de la hemostasia
1. Prostaciclina ( PGI2 ) – se sintetiza en el endotelio, vasodilatador potente y un inhibidor de la agregación plaquetaria. 2. Antitrombina III ( AT III ) – principal inhibidor de la trombina. 3. Proteína C es activada por la trombina en presencia de la trombomodulina. 4. Plasmina se forma a partir del plasminógeno
 – se sintetiza en el endotelio, vasodilatador potente y un inhibidor de la agregación plaquetaria. 2. Antitrombina III ( AT III ) – principal inhibidor de la trombina. 3. Proteína C es activada por la trombina en presencia de la trombomodulina. 4. Plasmina se forma a partir del plasminógeno.")
9
FISIOPATALOGÍA Es la clave para la comprensión de la clínica y biología de las enfermedades hemorrágicas y trombóticas

10
HISTORIA CLÍNICA Y LA EXPLORACIÓN FÍSICA
Son fundamentales para evaluar a los pacientes con síntomas y sin ellos. AHF – APP – Sangrado espontáneo de las mucosas petequias – púrpura – equimosis – Equimosis profundas – hematomas – hemartrosis – hemorragia tardía -

11
EL ESTUDIO DE LABORATORIO
Recuento y función plaquetaria Tiempo de sangría TTP – es el tiempo que tarda en formarse el coágulo. Mide la vía intrínsica. TP – es el tiempo que tarda en formarse el coágulo después de añadir tromboplastina( factor hístico y lipídico ). Mide la vía extrínsica
. Mide la vía extrínsica.")
12
EL ÍNDICE INTERNACIONAL NORMALIZADO
INR permite la comparación de las pruebas TP en diferentes laboratorios con reactivos tromboplastínicos distintos. Se emplea para vigilar el tratamiento con warfarina.

13
LOS INHIBIDORES Son ACS que interfieren en las pruebas de coagulación.
Los ACS específicos contra los factores por ejemplo: inhibidores de los factores VIII y IX. El ACL, es el inhibidor más común. Pro-longan el TTP y a veces el TP.

14
Estudios de la CID Estudios seriados para separar la CID de la insuficiencia hepática. Plaquetas – prolongación del TTP. Fibrinógeno – PDF – Dímeros D - Frotis en sangre periférica. Déficit del Factor XIII -

15
Estudio de Hipercoagubilidad
Trastornos Tromboembólicos Hereditarios: Actividad de la AT III Nivel y la actividad de la Proteína C. Nivel de la proteína S Nivel de la homocisteína. Actividad del Fibrinógeno

16
Recordar en el estudio Hipercoagublilidad
AT III durante el uso de heparina. Proteínas S y C antes de Warfarina Trombosis Aguda, anticonceptivos orales y la hepatopatía pueden reducir los niveles de estas tres proteínas. El embarazo < proteínas S y C. S. Nefrótico < AT III y > proteína C y S.

17
BALANZA HEMOSTÁTICA Para que la sangre pueda cumplir con sus múltiples funciones en la economía del organismo es necesario que se mantenga contenida en el territorio vascular y circulando libremente; así la sangre no se coagula, las plaquetas no se adhieren entre sí, ni al endotelio mientras no haya lesión vascular o exposición a alguna molécula procoagulante.

18
FASE VASCULAR Vasos lesión vascular activación plaquetaria
activación de la coagulación vasoconstricción

19
FASE PLAQUETARIA Agregados denominados tapones hemostáticos – materia prima sustancias producen vasoconstricción - Serotonina - Tromboxano A2 Inician la reparación de la pared vascular

20
FASE PLASMÁTICA Todos los factores se encuentran en forma de proenzimas y en el cual se dan una serie de reacciones logrando su activación y formando así un complejo activador de protrombina constituído por el factor: - Xa - Va y VIIIa - Fosfolípidos

21
FISIOLOGIA DE LA COAGULACIÓN
Formación activador de la Protrombina Tromboplastina Formación de Trombina Formación de Fibrina

22
Formación del Activador de la Protrombina
Se puede dar por dos mecanismos: Vía Intrínsica – requiere solo sustancias presentes en el plasma. Vía + lenta 4-6 m. Vía Extrínsica – requiere de un factor liberado por los tejidos que han sido lesio-nados, llamado Factor Activador Tisular y se desarrolla en seg. Vía + rápida.

23
Formación del Activador de la Protrombina
Lesión vascular – continuidad del endotelio y membranas, esto se acompaña de libera-ción de: Fosfolípidos Colágeno Factor activador tisular

24
COLÁGENO Produce A Plaquetaria, estas A se van a adherir entre sí y a la pared del vaso lesionado secretar + plaquetas, el factor V promueve la formación de: Trombina Serotonina vasoconstricción Tromboxano A2 Factor de crecimiento derivado de plaquetas

25
Inicia la regeneración de la pared vascular
Proporciona sitios en la superficie de membrana receptoras IIIa y IIb para la unión del Fibrinógeno y otras proteínas adhesivas para + agregación. En el interior de las plaquetas se activa un mecanismo de contracción de la actomiosina plaquetaria para consolidar el tapón .

26
Trastornos Vasculares
Síndrome de EHLERS – DANLOS Adquiridos: escorbuto – Síndrome o la Enfermedad de Cushing – Trastornos tóxico-metabólicos – Gamapatías monoclonales – Púrpura Senil – Congénitos: Purpura anular telangiectoide –Telengectasia hemorrágica hereditaria – Púrpura alérgica ( Shonlein-Henoch )
")
27
Trastornos Plaquetarios
Causas: 1. Formación defectuosa del tapón hemostático y hemorragias debido a la reducción de plaquetas: Trombocitopenia 2. Alteración de la función a pesar de la existencia de cifras normales de plaque-tas: Disfunción Plaquetaria.

28
TROMBOCITOPENIAS Pueden aparecer por tres mecanismos:
1. Menor producción de la médula ósea 2. Aumento del secuestro esplénico. 3. Destrucción acelerada de plaquetas.

30
Clasificación de las Trombocitopenias
PTI o Inmunológica. Trombocitopenias inmunológicas 2ª a : Hiperesplenismo HIV Post-transfusional Enfermedad del Colágeno Enfermedad Linfoproliferativa

31
Anomalías de la Función PLAQUETARIA
La hemostasia normal exíge 3 reacciones esenciales: 1. Adhesividad 2. Agregación 3. Desgranulación

32
Trastornos de la Adherencia Plaquetaria
A) Hereditarios: 1. Síndrome de Bernard-Soulier 2. Enfermedad de Von Willebrand B) Adquiridos: 1. Hiperazoemia 2. Enf. de Von Willebrand adquirida
 Hereditarios: 1. Síndrome de Bernard-Soulier. 2. Enfermedad de Von Willebrand. B) Adquiridos: 1. Hiperazoemia. 2. Enf. de Von Willebrand adquirida.")
33
Enfermedad de Von Willebrand
Se sintetiza en la célula endotelial y se almacena en los cuerpos de Weibel-Palade, y en los megacariocitos, alfaplaquetarios. La concentración FvW en plasma - 10mg/L. La < moderada del FvW o una pérdida selectiva de los procursores provoca menor adhesivad de las plaquetas seguida de hemorragias evidentes

34
FUNCIONES Favorece la adhesivad de las plaquetas
Actúa como portador del Factor VIII, ( Factor Antihemofílico ) una proteína esencial para la coagulación de la sangre
 una proteína esencial para la coagulación de la sangre.")
35
DEFINICIÓN Enfermedad hemorrágica donde el defecto primario es una reducción de la síntesis o una anomalía química de un antígeno relacionado con el factor VIII lo que ocasiona disfunción plaquetaria

36
INCIDENCIA Autosómica dominante Relativamente frecuente
Afecta ambos sexos 1: 800 – 1000 individuos Puede registrarse historia materna o paterna ( + ).
.")
37
Manifestaciones Clínicas
Epistaxis frecuentes o sangrados por muco-sa bucal, gastrointestinal o genitourinaria. Facilidad para la formación de hematomas. Metrorragias – Menorragias Hemorragias por cortes cutáneos. Hemorragias posterior a intervenciones quirúrgicas ( extracciones dentales ).
.")
38
LABORATORIO T.S. Prolongado
< de la concentración en plasma del FvW. < de la actividad biológica medida con el análisis del cofactor ristocetina. < de la actividad del factor VIII.

39
TRATAMIENTO Crioprecipitados ( derivado del plasma rico en factor VIII ). 10 bolsas bid cada 48 – 72 hrs.

40
Clasificación de las Trombocitopenias
Trombocitopenias inducidas por fármacos: Heparina Trombocitopenia, eritrocitos fragmentados y hemólisis: PTT Embolias tumorales metastásicas Síndrome Hemolítico Urémico - SHU

41
Trastornos de Agregación Plaquetaria
A) Hereditarios 1. Tromboastenia de Glanzmann 2. Afibrinogemia B) Adquiridos 1. Inhibición de los PD de Fibrina 2. Disproteinemias 3. Consumo de Fármacos
 Hereditarios. 1. Tromboastenia de Glanzmann. 2. Afibrinogemia. B) Adquiridos. 1. Inhibición de los PD de Fibrina. 2. Disproteinemias. 3. Consumo de Fármacos.")
42
Trastornos de la liberación de las Granulaciones
A) Hereditarios: 1. Albinismo Oculo-cutáneo 2. Sd. de Chediak – Higashi 3. Déficit aislado de granulaciones B) Adquiridos: 1. Derivación cardiopulmonar 2. Trastornos mieloproliferativos 3. Fármacos como Aspirina y otros AINES.
 Hereditarios: 1. Albinismo Oculo-cutáneo. 2. Sd. de Chediak – Higashi. 3. Déficit aislado de granulaciones. B) Adquiridos: 1. Derivación cardiopulmonar. 2. Trastornos mieloproliferativos. 3. Fármacos como Aspirina y otros AINES.")
43
Trastornos de la Coagulación
Hereditarios: Hemofilia A. Hemofilia B. Adquiridos: Déficit de vitamina K Hepatopatías CID

44
HEMOFILIA A Es la coagulopatía más común.
Enf. recesiva ligada al cromosoma x. Causa: deficiencia o ausencia del factor de coagulación VIII plasmático. Por tanto, según el nivel de actividad de estos factores, se habla de hemofilia severa, moderada o leve. Puede ser por mutación espontánea o proceso inmunológico adquirido.

45
FISIOPATOLOGÍA niveles de F. VIII, o
este factor disfuncional, o inhibidores del factor VIII, llevan a la disrupción de la cascada intrínsica de la coagulación. Lleva a la formación del activador de la protrombina o tromboplastina intrínsica, por lo que ésta se forma lentamente.

46
Sintomatología y sitios de hemorragia
Dominada por la hemorragia. Puede aparecer precozmente desde los primeros días de vida. Sitios de hemorragia: Articular – SNC – Mucosas – Gastrointestinal – Genitourinario – Pulmonar – Cardiovascular.

47
Frecuencia y Formas de Hemofilia A
1/ – varones nacidos tienen hemofilia A. Enfermedad Severa: con < de 1% de actividad del FVIII, presente en menores de un año, en el 70% casos. Enfermedad Moderada: se define p/ 1-5% de actividad del FVIII, presente a edades de uno-dos años, en el 15% casos.

48
Formas de Hemofilia A Enfermedad leve: con + del 5% de actividad del FVIII, presente en mayores de 2 años y representa el 15% casos. La hemofilia esporádica: que se presenta en un 30% de los niños hemofílicos, que no tienen ningún antecedente familiar.

49
Estudios de Laboratorio
Hb – Hto – Plaquetas - TP: mide pasos de la vía extrínsica. TPT: screen de los pasos intrínsicos. Factor VIII: nl 50 – 150% Severo: mayor a 1%, Moderado: 1 – 5% y Leve: menor a 5%. Inhibidores del FVIII: 0 a 10 unidades.

50
HEMOFILIA B Desorden hereditario ligado a x que re- sulta de una disminución en la activ. del factor IX. ( K dependiente ). Enfermedad de Chistmas. Incidencia 1: Manifestaciones clínicas iguales a la HA Aparece solo en el sexo masculino.

51
Fisiopatología Bajos niveles de Factor IX, factor disfun-cional, inhibidores llevan a la disrupción de la cascada de la coagulación intrínsica, con resultado de hemorragia. La clínica y el laboratorio son similares a los de la Hemofilia A. También las compli-caciones del sangrado o infecciones.

52
FORMAS DE HEMOFILIA B Severa: < de 1% de actividad del factor. Se encuentra en un 50% de los casos. Presente en < de 1 año. Moderada: 1 – 5 % de actividad, en un 30% de los casos, 1 – 2 años presente- Leve: > de 5% de actividad, 20% de los casos, en mayores de 2 años presente.

53
Manifestaciones Clínicas
Hemorragias internas durante toda la vida primer episodio antes de los 18 meses. Traumatismos mínimos pueden originar hemorragias tisulares extensas y hemartrosis

54
Datos de Laboratorio TTP prolongado TS normal TP normal
Las determinaciones específicas del factor VII y IX determinan el tipo y gravedad de la hemofilia

55
Trastornos Adquiridos de la Coagulación
1.Déficit de Vitamina K 2. Hepatopatías 3. CID 4. Anticoagulantes Circulantes

56
Déficit de Vit K. El hepatocito necesita de la vit k. Para sintetizar los factores: II – VII – IX – X – La vit k. Puede faltar por muchas razones: 1. Drogas que tienen efecto antagonista. 2. No se absorbe, es liposoluble. 3. Fuente exógena, contenida en los animales y la endógena, sintetizada en la flora bacteriana intestinal.

57
HEPATOPATÍAS Pueden alterar la hemostasia a través de la síntesis alterada de los factores de la coagulación. Aumento de la fibrinolisis. Trombocitopenia asociada. Hígado graso agudo del embarazo, consumo de factores por una CID.

58
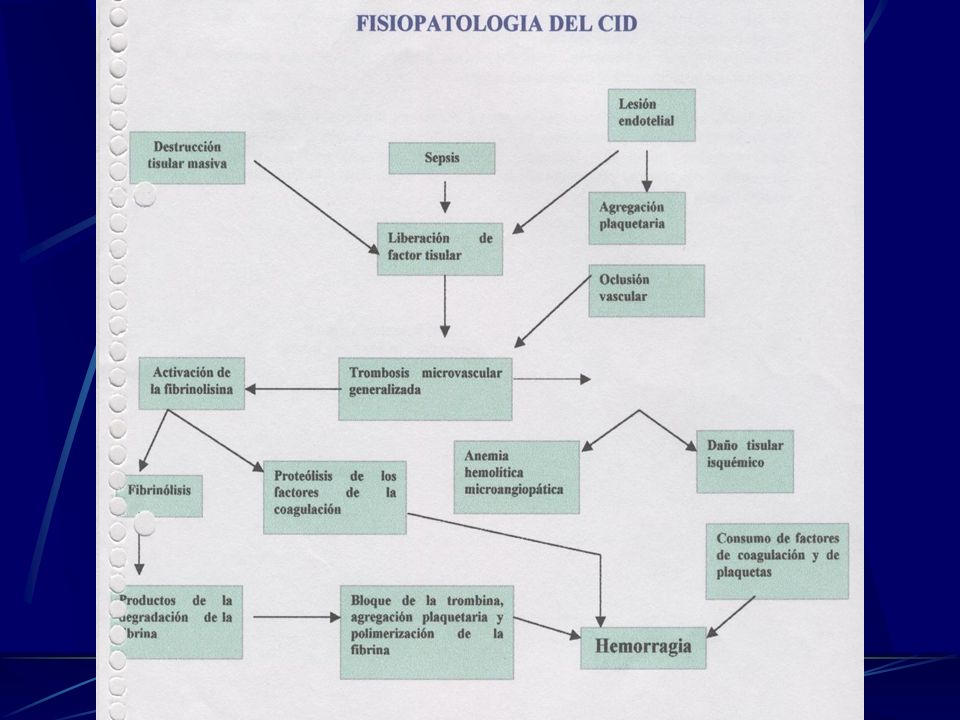
Coagulación Intravascular Diseminada
Trastorno tromboembólico que puede ser: agudo - subagudo o crónico secundario. Se caracteriza por: 1. Activación de la cascada de la Coagulación. 2. Mecanismos de Fibrinolisis.

59
CID Se puede manifestar por:
Signos y Síntomas de hipoxia tisular. Trastornos hemorrágicos Etiológicamente se concluye que la CID se debe a una activación anormal de las vías intrínsica y extrínsica de la coagulación y/o alteración de las influencias supresoras de la coagulación

60
Factores que estimulan liberación del Factor Tisular
Placenta Gránulos de las células leucémicas Moco de ciertos adenocarcinomas Endotoxinas bacterianas Monocitos que liberan IL 1 y FNTą que estimulan el factor Tisular, también < producción de trombomodulina ( activa la proteína C anticoagulante).
.")
61
Factores que producen lesión endotelial
Lesión mecánica Lesión endotelial difusa ( depósitos complejos Autoinmunes LES. Temperaturas extremas Microorganismos ( rickettsias meningo ) Pueden combinarse los factores. Consecuencias del CID
 Pueden combinarse los factores. Consecuencias del CID.")
63
Anticoagulantes del CID
Son sustancias endógenas que inhiben la coagulación de la sangre. En general se trata de unos Acs, que neutralizan la actividad de un factor de la coagulación. O la actividad de un fosfolípidoprocoagu-lantes empleado en ciertas bacterias de pruebas de la coagulación ( ACL ).
.")
64
RESUMEN Está dividida en las tres fases: Hemostasis 1. Fase vascular
2. Fase plaquetaria 3. Fase plasmática Fisiología de la coagulación: 1. En la formación del Act. de la Protrombina 2. Protrombina en Trombina 3. Transformación de Fibrinógeno en Fibrina

65
RESUMEN Factor intrínseco y extrínsico, ambos son necesarios en condiciones fisiológicos para que se verifique una hemostasis . La deficiencia en uno de ellos se manifiesta en la clínica con un trastorno hemorrágico, es decir no se sustituyen, sino que se complementan.

66
RESUMEN La diferencia entre ellos es que el mecanismo intrínseco necesita sólo de sustancias presentes en el plasma y es más lenta ( 4 – 6 minutos ), mientras que el mecanismo extrínsico necesita de un factor liberado por los tejidos lesionados y es más rápida ( 10 – 13 segundos ).
, mientras que el mecanismo extrínsico necesita de un factor liberado por los tejidos lesionados y es más rápida ( 10 – 13 segundos ).")
67
RESUMEN Mecanismo intrínseco viene estudiado con el TTP activado y por esto sus valores son de 30 – 45 segundos. Mecanismo extrínsico con el TP y es por esto sus valores son de 12 – 15 sgs 65 – 100%

68
RESUMEN Trastornos hemorrágicos por vascular.
Trastornos hemorrágicos por plaquetas. Coagulopatía. TS: detecta la fase vascular y plaquetaria - TP: estudia la f. Intrínsica de la coagulación. TTP: estudia la f. Extrínsica de la coagulación

69
Estructura de la Plaqueta

70
¿ Qué es la hemostasia ? Los vasos sanguíneos, las plaquetas y los factores de la coagulación trabajan de forma sincronizada para conseguir el objetivo común de impedir la salida de la sangre de los vasos sanguíneos, a este objetivo se le conoce como hemostasia.

71
¿Cuáles son los mecanismos de la hemostasia?
Cualquiera de los mecanismos implicados en la hemostasia puede verse alterado dando lugar a problemas de sangrado que se manifiesten como enfermedades hemorrágicas. Para comprender las enfermedades hemorrágicas es preciso tener unas nociones de hemostasia básicas.

72
La hemostasia se entiende mejor si se divide en 3 componentes:
Fase Vascular Fase Plaquetaria Fase Plasmática

73
Fase Vascular La pared del vaso sanguíneo está recubierta por células endoteliales que separan las células sanguíneas y el plasma del tejido circundante apoyadas en una matriz subendotelial. Permiten el intercambio de gases, sustancias nutritivas y el paso de algunas células.

74
Fase Vascular Las células endoteliales producen sustancias que impiden la agregación de las plaquetas. Los nervios y el tejido muscular que rodea al vaso permiten el cierre del vaso cuando se lesiona. La constricción del vaso frena o detiene el flujo sanguíneo de forma temporal impidiendo el sangrado.

75
Fase Plaquetaria Las plaquetas son fragmentos celulares con forma de disco ovoide que se asemejan a una esponja. Están formadas por una zona periférica, una zona central y diversos gránulos. La zona periférica esta formada por la membrana y región subyacente. Contiene múltiples receptores que le permiten captar los estímulos y poner en marcha las reacciones de adhesión y agregación. La zona central está formada por fibras de distinto tipo que le permiten mantener la forma y realizar la contracción plaquetaria. Se distinguen gránulos alfa, densos y lisosomas.

76
Fase Plaquetaria Se distinguen gránulos alfa, densos y lisosomas. Los gránulos almacenan productos que se liberan en el momento adecuado aportando energía y favoreciendo la agregación para formar el tapón plaquetar. La función de la plaqueta es formar el coagulo provisional o tapón plaquetario y contribuir a la formación del definitivo aportando fosfolípidos.

77
Fase Plaquetaria Para su funcionamiento adecuado requieren de otros factores conocidos como cofactores. Por último, el fibrinógeno, una proteína plasmática, es la diana final de los factores coagulantes. La acción de los factores activados final es la transformación del fibrinógeno soluble en fibrina. El resultado final es la formación del coagulo de fibrina, que es frágil y requiere ser reforzado por el factor XIII. Se requiere de fosfolípidos y calcio para todo el proceso.

78
Fase Plasmática El proceso depende de la activación de varias proteínas que circulan en la sangre en forma inactiva conocidas como factores de la coagulación. Estas proteínas se producen en su mayor parte en el hígado. Algunas de ellas precisan de vitamina K para fabricarse de modo que funcionen correctamente. Durante el proceso de coagulación los factores de la coagulación se activan en forma secuencial influyendo unos sobre otros.

79
¿A que llamamos enfermedades o trastornos hemorrágicos?
Las enfermedades hemorrágicas son todos aquellos procesos generales en los cuales se observa una tendencia en mayor o menor grado al sangrado sin una causa local clara que lo justifique. Las enfermedades hemorrágicas son la consecuencia del funcionamiento defectuoso de los mecanismos que mantienen el flujo sanguíneo dentro del sistema vascular. Por tanto, las enfermedades hemorrágicas se deben a un mal funcionamiento de la hemostasia cuyo origen puede ser muy variado.

80
¿Qué produce las enfermedades hemorrágicas?
Las enfermedades hemorrágicas no tienen una causa común. Son muchos los factores causales que pueden intervenir en cada uno de los procesos involucrados en la hemostasia.

81
Estructura Vascular La debilidad o anomalía de la estructura vascular impide la respuesta normal del vaso y condiciona una púrpura vascular. La deficiencia de vitamina C, la inflamación, el daño inmunológico, ciertas toxinas, el envejecimiento y algunos defectos congénitos pueden afectar a la pared vascular. Ello origina fragilidad de la misma y salida de la sangre de los vasos, que a nivel de la piel origina la lesión llamada púrpura.

82
Fase Plaquetaria La disminución en el número de plaquetas puede llevar a hemorragia. Las plaquetas pueden disminuir por: a) falta de producción de la medula ósea, sustitución de la medula por tumor o enfermedad maligna, daño por fármacos, infecciones o mecanismo inmunológico-, b) anormal fabricación por la medula en mielodisplasias y anemias megaloblásticas, c) aumento de la destrucción o utilización de las plaquetas, d) almacenamiento de plaquetas en el bazo aumentado de tamaño.
 falta de producción de la medula ósea, sustitución de la medula por tumor o enfermedad maligna, daño por fármacos, infecciones o mecanismo inmunológico-, b) anormal fabricación por la medula en mielodisplasias y anemias megaloblásticas, c) aumento de la destrucción o utilización de las plaquetas, d) almacenamiento de plaquetas en el bazo aumentado de tamaño.")
83
Fase Plaquetaria También un funcionamiento anormal de las plaquetas impide la formación del tapón plaquetario. Las plaquetas pueden funcionar mal en un momento determinado por: a) medicamentos -aspirina, antiinflamatorios no esteroideos, etc.-, b) cifras muy elevadas de plaquetas anómalas en síndromes mieloproliferativos, c) paraproteínas.
 medicamentos -aspirina, antiinflamatorios no esteroideos, etc.-, b) cifras muy elevadas de plaquetas anómalas en síndromes mieloproliferativos, c) paraproteínas.")
84
Fase Plaquetaria En otros casos, las alteraciones son congénitas secundarias a anomalías en estructuras necesarias para la función plaquetaria. La púrpura es una lesión típica del mal funcionamiento o escaso número de plaquetas. Las lesiones de tipo púrpura puntiforme se llaman petequias, y las más extensas como placas equimosis.

85
Fase Plasmática La ausencia de factores de la coagulación necesarios para que tenga lugar la coagulación de la sangre impide que se forme el coagulo de fibrina y conduce a hemorragia. La alteración en el funcionamiento de estos factores de origen congénito o adquirido provoca sangrado de forma similar. La deficiencia congénita de mayor importancia por su frecuencia es la hemofilia A por déficit de actividad del factor VIII.

86
Fase Plasmática El déficit de factor IX se llama hemofilia B. La deficiencia de vitamina K conduce a una síntesis anormal de varios factores que ocasiona tendencia al sangrado. El consumo de factores por la activación mantenida de la coagulación conduce a unos niveles disminuidos. Este proceso se asocia a infecciones, complicaciones del embarazo, carcinomas metastásicos, leucemia aguda- sobretodo promielocítica-, daño tisular severo, reacción transfusional por incompatibilidad ABO y mordeduras de serpientes.

87
¿Cuáles son los datos clínicos a conocer sobre las enfermedades hemorrágicas?
Los antecedentes previos de hemorragias y hematomas son sugestivos de un trastorno de la hemostasia. El tiempo durante el cual se llevan padeciendo orienta a un proceso congénito o adquirido. La frecuencia de los hematomas y el tamaño son importantes.

88
¿Cuáles son los datos clínicos a conocer sobre las enfermedades hemorrágicas?
El sangrado con pequeños cortes y con intervenciones quirúrgicas o extracciones dentales. En las mujeres se deben conocer las características del sangrado menstrual. La existencia de sangrados graves previa puede apoyar.

89
¿Cuáles son los datos clínicos a conocer sobre las enfermedades hemorrágicas?
La existencia de familiares con anomalías en la coagulación apoya un origen heredado. El sexo de las personas en la misma familia afectadas también es importante, por ejemplo, la hemofilia afecta casi exclusivamente a varones. Algunos fármacos pueden inducir alteraciones de la hemostasia, bien de las plaquetas o de la coagulación como heparina y anticoagulantes orales.

90
¿Cuáles son los datos clínicos a conocer sobre las enfermedades hemorrágicas?
Hay que descartar la existencia de enfermedades que producen una alteración del sistema hemostático: las enfermedades hepáticas al no fabricarse los factores de la coagulación de forma normal; en las enfermedades renales se producen interferencias con la función de las plaquetas y otros procesos de la coagulación que facilitan el sangrado.

Presentaciones similares











>")
 es una alteración fisiopatólogica sistémica, trombohemorrágica, que se presenta en algunas situaciones.>")







