Descargar la presentación
La descarga está en progreso. Por favor, espere

1
HEMOGLOBINA y hemoglobinopatias
Mary l. vallecillo. MsC

2
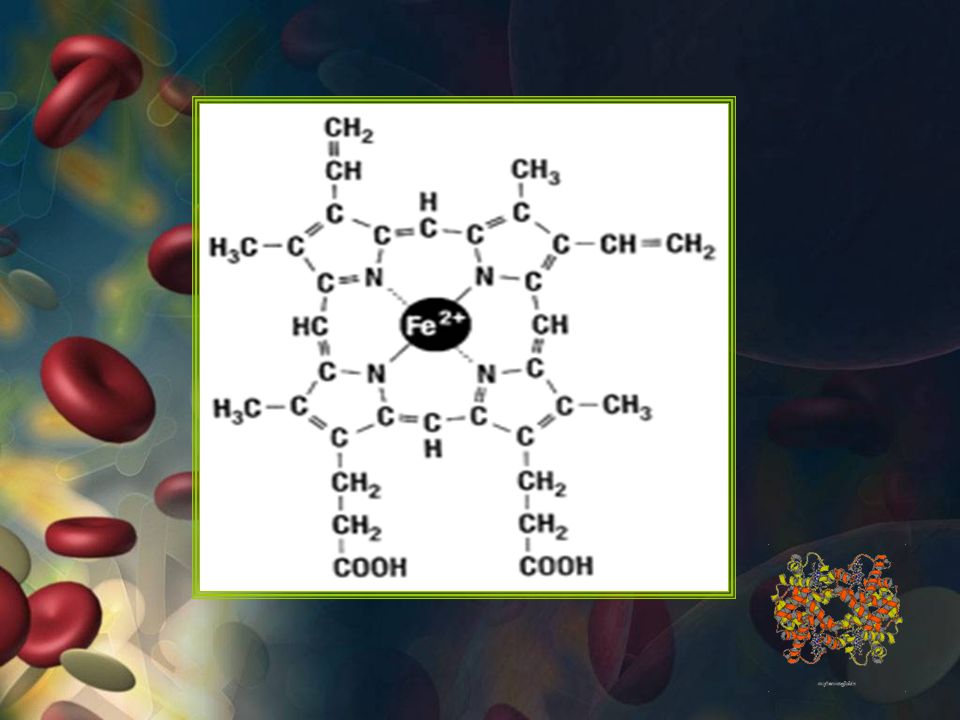
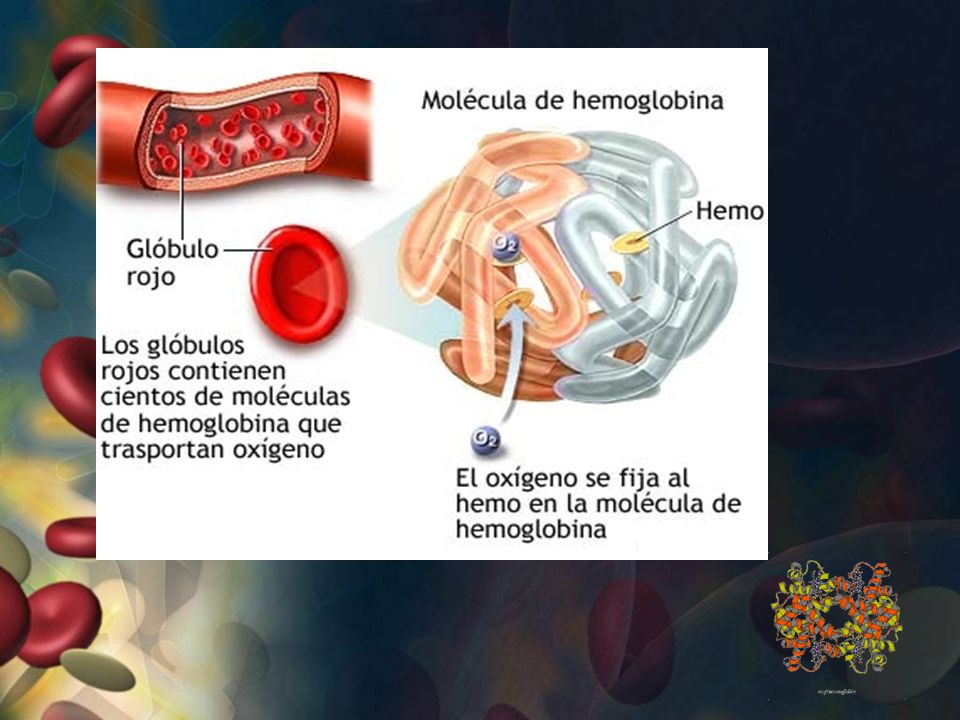
HEMOGLOBINA Proteína globular Presente en glóbulos rojos
transporta O2 -CO2 y H+ Esférica y 6,4 nm diámetro 4 cadenas polipeptidicas de globina - Hemo 13 – 18 g/ dl en el hombre 12 – 16 g/dl en la mujer.

4
HEMOGLOBINA Características son: Transporte oxígeno
Capacidad de “captar” y “ceder “ oxígeno Capacidad de amortiguar una solución de bicarbonato Gran solubilidad

6
HEMOGLOBINA Tipos de hemoglobina: Hb adulta- HbA1: 2 y 2 (96%)
Hb fetal- HbF: 2 y 2 (1%)
")
7
HEMOGLOBINA Estados de la Hb: Oxihemoglobina:
rojo intensos *sangre arterial rojo oscuro *sangre venosa Dexosihemoglobina: -Hb reducida “T” o tensa

9
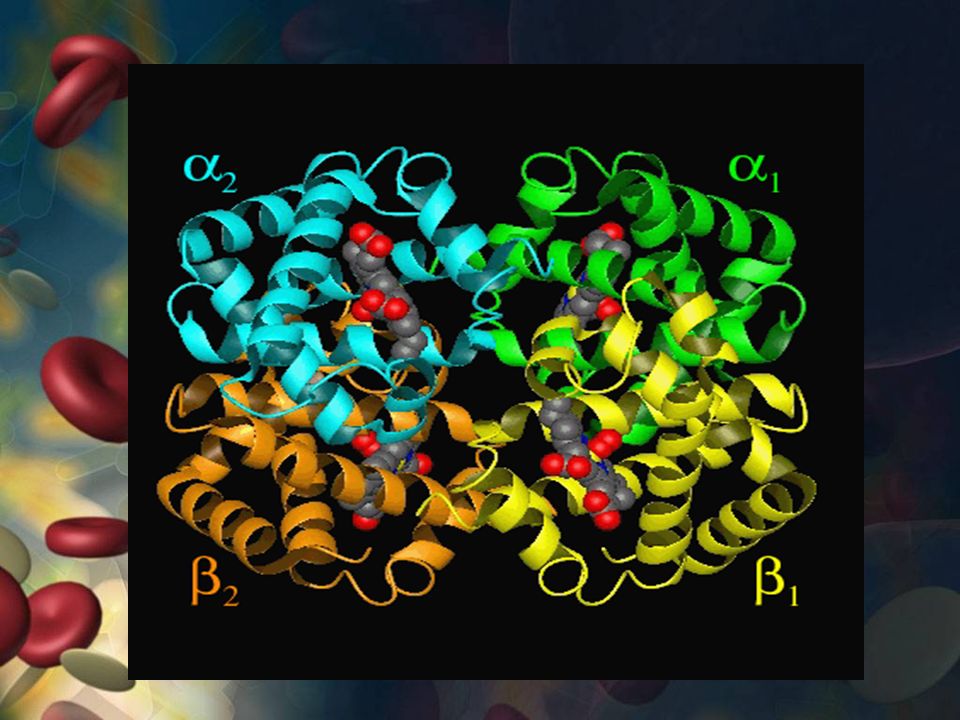
HEMOGLOBINA Estructura de la Hb: Proteína cuaternaria
4 cadenas polipeptídicas Dimeros (alfa-beta)1 y (alfa-beta)2 Unidas por interacciones hidrofóbicas
1 y (alfa-beta)2. Unidas por interacciones hidrofóbicas.")
11
HEMOGLOBINOPATIAS Defecto de carácter hereditario
Tiene como consecuencia una estructura anormal en una de las cadenas de las globina Simple cambio de un aminoácido en una de las cadenas de globina Afectación de la cadena beta son más frecuentes

12
CLASIFICACION 1. Hemoglobinopatías Estructurales * Hb S, Hb C, Hb E
2. Talasemias: * Alfa, Beta 3. Variantes de la Hb talasémicas * Hb E 4. Persistencia hereditaria de la Hb fetal 5. Hemoglobinopatias adquiridas * Metahemoglobinemia * Sulfohemoglobina

13
HEMOGLOBINA S Anemia Falciforme o Drepanocítica. Pauling, 1949
Enfermedad hereditaria, autosómica recesiva Raza negra La anomalía se sitúa en la cadena beta, cuya glutamina es sustituida por valina

14
HEMOGLOBINA S Cambio en el codón GAC normal que pasa a GTG
Glóbulo rojo en forma de hoz

15
HEMOGLOBINA S vaso-oclusión Flujo de sangre lento
Adherencia de los eritrocitos al endotelio La desoxigenación de las células produce salida de potasio de los glóbulos rojos, lo cual aumenta la densidad de los glóbulos y la tendencia de la HbS a polimerizarse El componente hemo tiende a liberarse de la proteína debido a la polimerización de la hemoblobina S.

16
HEMOGLOBINA C Se caracteriza por la sustitución del ácido glutámico de la cadena beta por lisina El estado homocigoto (CC) se caracteriza por una ligera anemia hemolítica crónica con esplenomegalia; la vida media del eritrocito esta disminuida El estado heterocigoto (AC) no produce trastorno alguno.
 se caracteriza por una ligera anemia hemolítica crónica con esplenomegalia; la vida media del eritrocito esta disminuida. El estado heterocigoto (AC) no produce trastorno alguno.")
17
Sus síntomas incluyen un mayor riesgo de que se produzcan infecciones y también periodos dolorosos y un bazo agrandado No tiene cura; pero hay tratamientos para los problemas que provoca

18
HEMOGLOBINA E Resulta de una mutación en la cadena beta de la hemoglobina. Las personas con enfermedad de la hemoglobina E tiene una anemia hemolítica suave y una esplenomegalia suave. El rasgo de la hemoglobina E es benigno

19
TALASEMIA Enfermedad hereditarias de la sangre
Defecto en las cadenas alfa y beta. Anemias hemolíticas -microcitosis (disminución del tamaño) hipocromia (disminución del color) Eritrocitos blancos
 -hipocromia (disminución del color) -Eritrocitos blancos.")
20
TALASEMIA Se dividen en: 1.Alfa talasemias 2.Beta talasemias

21
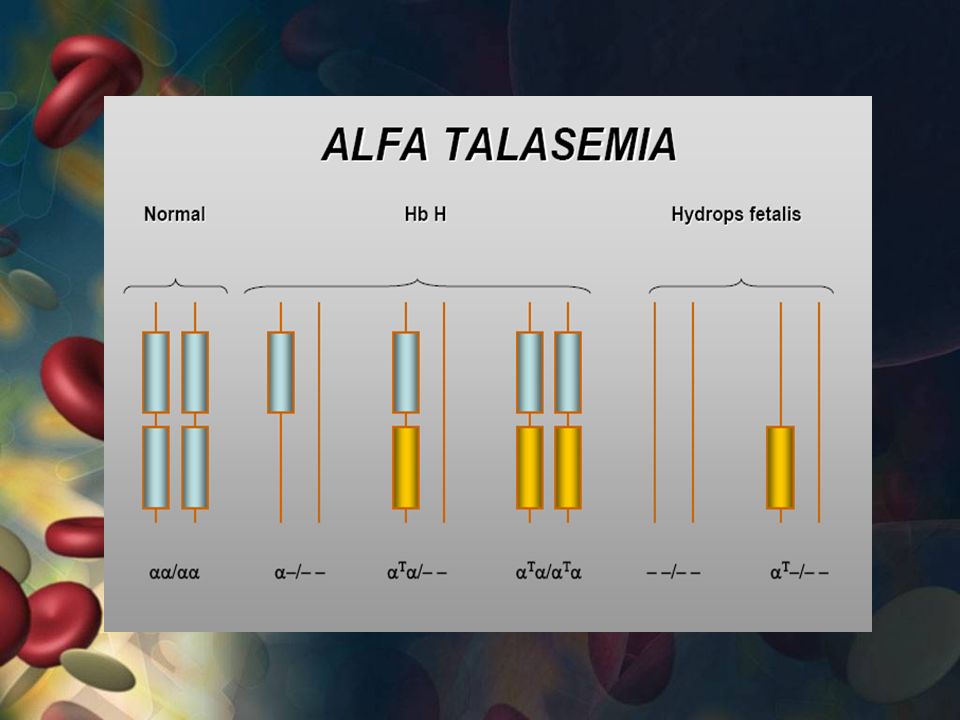
TALASEMIA ALFA Hay mutaciones en la cadena alfa de la hemoglobina
2 genes de la cadena alfa en cada cromosoma 16 (4 en total) Tipos de talasemia alfa: 1. El portador silencioso 2. Talasemia alfa menor 3. Hemoglobina H 4. Talasemia alfa mayor
 Tipos de talasemia alfa: 1. El portador silencioso. 2. Talasemia alfa menor. 3. Hemoglobina H. 4. Talasemia alfa mayor.")
22
EL PORTADOR SILENCIOSO
Más leve Solo gen de globina alfa anormal o faltante. No síntomas Pueden transmitir

23
TALASEMIA ALFA MENOR 2 genes de globina alfa anormales o faltantes.
No síntomas Anemia leve Transmitir la enfermedad a sus hijos.

25
HEMOGLOBINA H 3 genes de globina alfa faltantes o anormales
Anemia de leve a moderada. -Infecciones virales -Medicamentos sulfa Agrandamiento del bazo y cálculos biliares.

26
TALASEMIA ALFA MAYOR Más grave No hay genes.
Anemia grave, insuficiencia cardíaca y acumulación de líquidos. Nacen sin vida o mueren pocas horas después del parto. Transfusiones de sangre de por vida

28
TALASEMIA BETA Causada por mutaciones en la cadena beta de la molécula de hemoglobina. Existe un gen para la cadena beta en cada cromosoma número 11, con un total de dos genes. 3 formas principales: La talasemia Leve La talasemia Intermedia La talasemia Grave

29
TALASEMIA BETA

30
1. LA TALASEMIA MENOR Denominada rasgo talasémico
Causada por una mutación en un gen de globina beta. La mayoría de las personas afectadas no presenta síntomas aunque algunas padecen anemia leve. Las personas afectadas pueden transmitir el gen anormal a sus descendientes.

31
2. LA TALASEMIA INTERMEDIA
Resultado de anomalías en ambos genes de globina beta. Son menos graves que las que causan la talasemia mayor. Los niños afectados padecen anemia de leve a moderada , presentan agrandamiento del bazo y anomalías en los huesos. Necesitan transfusiones de sangre para reducir las complicaciones.

32
3. LA TALASEMIA MAYOR Forma más grave
Resultado de mutaciones graves en ambos genes de globina beta. También se denomina “anemia de Cooley”. La mayoría de los niños afectados parecen saludables al nacer. No obstante, durante el primer o el segundo año de vida se vuelven pálidos e irritables y pierden el apetito. Su crecimiento es lento y a menudo tienen ictericia (su piel y sus ojos adquieren un color amarillento).
.")
33
Muchas gracias por su atención

Presentaciones similares