Descargar la presentación
La descarga está en progreso. Por favor, espere

1
Dr. Luis A. Mora B. Cátedra de Bioquímica UCIMED
Proteínas y transporte de O2 Hemoglobina y mioglobina (Relación estructura:función) Dr. Luis A. Mora B. Cátedra de Bioquímica UCIMED
 Dr. Luis A. Mora B. Cátedra de Bioquímica. UCIMED.")
2
Introducción La función de las proteínas depende de su estructura y de los cambios conformacionales que puedan llevar a cabo. Ejemplo: Proteínas de transporte de oxígeno: mioglobina y hemoglobina

3
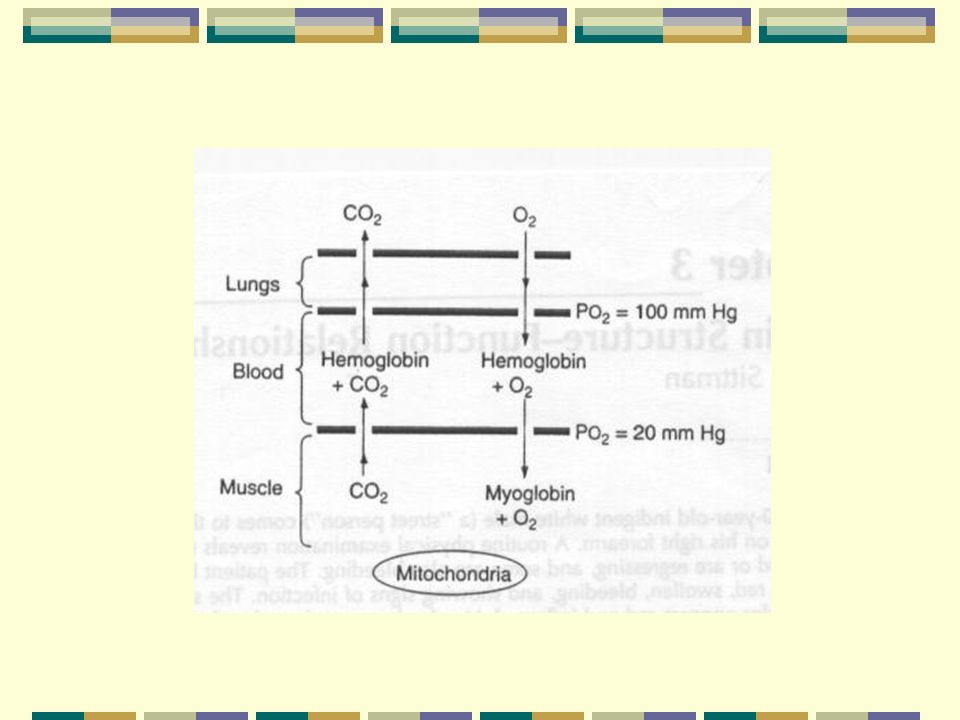
Pulmones Sangre Músculo

4
Pigmentos respiratorios:
Hemoglobina y citocromos ( rojos )-conjugadas. Grupo prostético: HEME : tetrapirrol cíclico naturaleza porfirínica Hemoproteínas: mioglobina,catalasa,peroxidasas, triptofano pirrolasa y xantina oxidasa. Cuatro cadenas polipeptídicas: estructura primaria origina propiedades fisicoquímicas y biológicas distintas. Estructura cuaternaria-forma tetraédrica.
-conjugadas. Grupo prostético: HEME : tetrapirrol cíclico. naturaleza porfirínica. Hemoproteínas: mioglobina,catalasa,peroxidasas, triptofano pirrolasa y xantina oxidasa. Cuatro cadenas polipeptídicas: estructura primaria origina propiedades fisicoquímicas y biológicas distintas. Estructura cuaternaria-forma tetraédrica.")
5
Mioglobina 1- Función: Células musculares
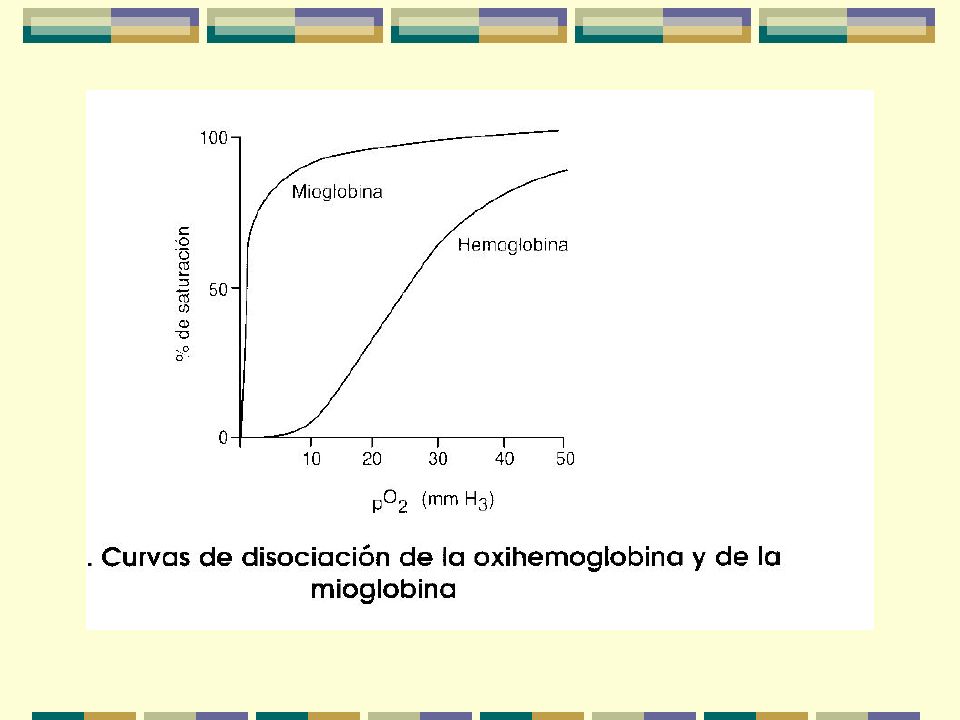
Almacena O2 y lo transporta a mitocondrias Tensión baja de O2 Curva de disociación de O2

7
Mioglobina 2- Estructura: -globina
una cadena de 153 residuos de AA aproximadamente el 75% de su estructura -posee 8 segmentos alfa helicoidales (A-H) -Residuos polares en el exterior y no polares en el interior. -heme: Se acomoda en bolsa hidrofóbica Función: unión del O2 a la Mb ( Hb). Sin heme no hay unión. Estructura: hierro ferroso (unión) y proto IX (4 grupos pirrólicos) Unión al oxígeno Valencias de coordinación o ligandos. Protección de la bolsa hidrofóbica contra la oxidación del Fe. Metamioglobina no funcional (H2O) Electrón extra impide formación de ligando para unión de O2
 -Residuos polares en el exterior y no polares en el interior. -heme: Se acomoda en bolsa hidrofóbica. Función: unión del O2 a la Mb ( Hb). Sin heme no hay unión. Estructura: hierro ferroso (unión) y proto IX (4 grupos pirrólicos) Unión al oxígeno. Valencias de coordinación o ligandos. Protección de la bolsa hidrofóbica contra la oxidación del Fe. Metamioglobina no funcional (H2O) Electrón extra impide formación de ligando para unión de O2.")
8
En rojo se representa el grupo Heme y en verde la cadena lateral de la histidina
a hélice se muestra como cintas rojas Cada bola roja es un átomo de hierro

9
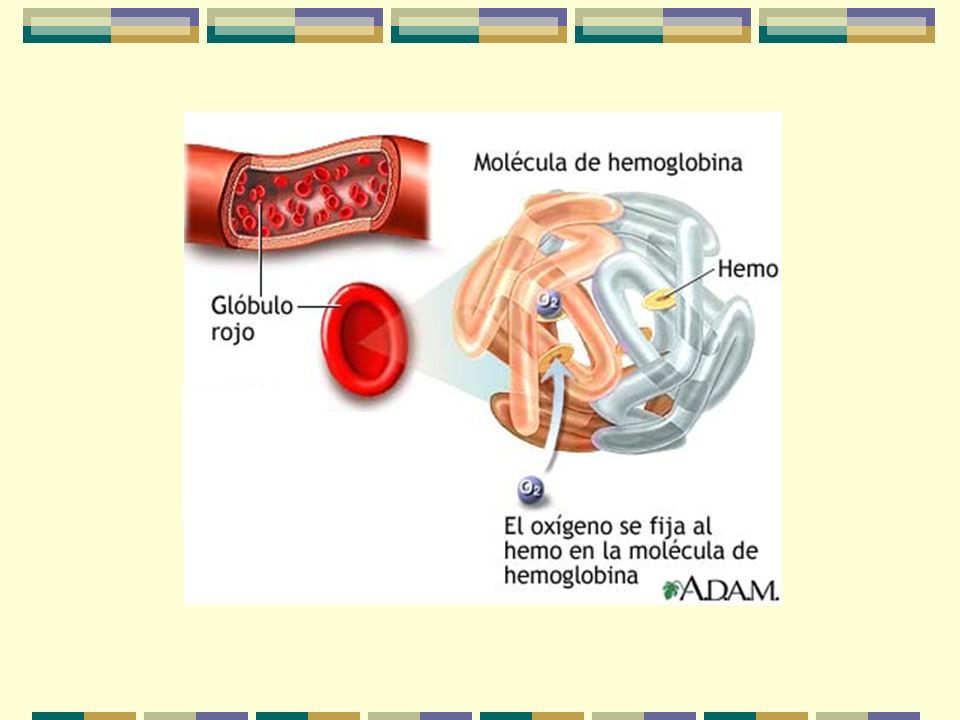
Heme Oxígeno Globina Estructura

11
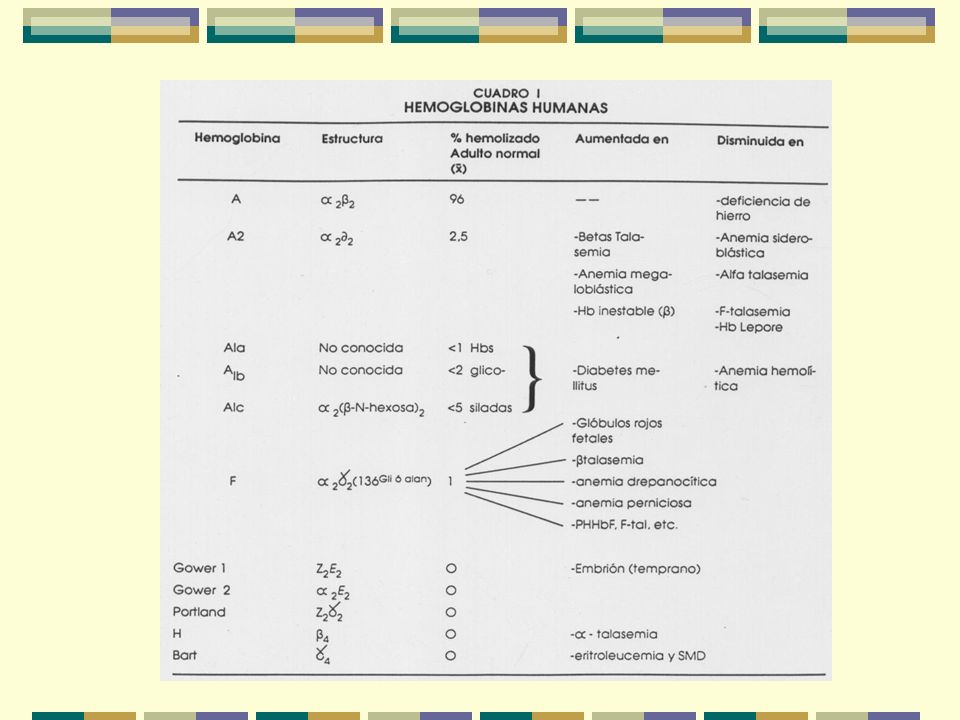
Hemoglobina 1- Características:
Su empacamiento le permite estar a altas conc. dentro del glób. rojo sin problemas de presión osmótica o viscosidad Hay 5 mil millones de glób. rojos/mL de sangre Cada glób. rojo tiene 280 millones de moléculas de Hb 2- Funciones: Capta O2 a altas PO2 en los pulmones Capaz de liberarlo a bajas PO2 Transporta CO2 de los tejidos a pulmones

12
Síntesis de la Hemoglobina

13
Heme Oxígeno Globina Estructura

14
a

15
En rojo se representa el grupo Heme y en verde la cadena lateral de la histidina
a hélice se muestra como cintas rojas Cada bola roja es un átomo de hierro

16
Diferencia funcional entre la hemoglobina y la mioglobina:
Curva de disociación Sigmoidal (Hb) vrs hipérbole (Mb) Unión del O2 es cooperativa en la Hb
 vrs hipérbole (Mb) Unión del O2 es cooperativa en la Hb.")
17
3- Estructura: Tetramérica
El Heme está contenido en bolsa hidrofóbica y es idéntico al de la Mb Presenta gran diversidad estructural y genética (según: edad y necesidades de O2)
")
19
Gower 1 (z2, e2) Gower 2 (a2, e2) Portland (z2, g2) Fetal (a2, g2) A (a2, b2) A2 (a2, d2)
 Gower 2 (a2, e2) Portland (z2, g2) Fetal (a2, g2) A (a2, b2) A2 (a2, d2)")
20
4- Relaciones función-estructura:
H+, 2-3 DPG y CO2 como efectores alostéricos, refuerza el concepto de esta relación Hb Fetal a- Unión cooperativa del O2 - DesoxiHb: cada unidad unida por enlaces electrostáticos ( el Fe está 0.4A fuera del plano del heme por repulsión estérica entre histidina proximal y los átomos de nitrógeno del anillo porfirínico). Forma T: baja afinidad por O2 - Oxi Hb: rompe los enlaces y origina cambios estructurales en donde los sitios quedan expuestos. Forma R: Mov. del Heme constante de afinidad: X500
. Forma T: baja afinidad por O2. - Oxi Hb: rompe los enlaces y origina cambios estructurales en donde los sitios quedan expuestos. Forma R: Mov. del Heme constante de afinidad: X500.")
21
Forma oxigenada y desoxigenada

22
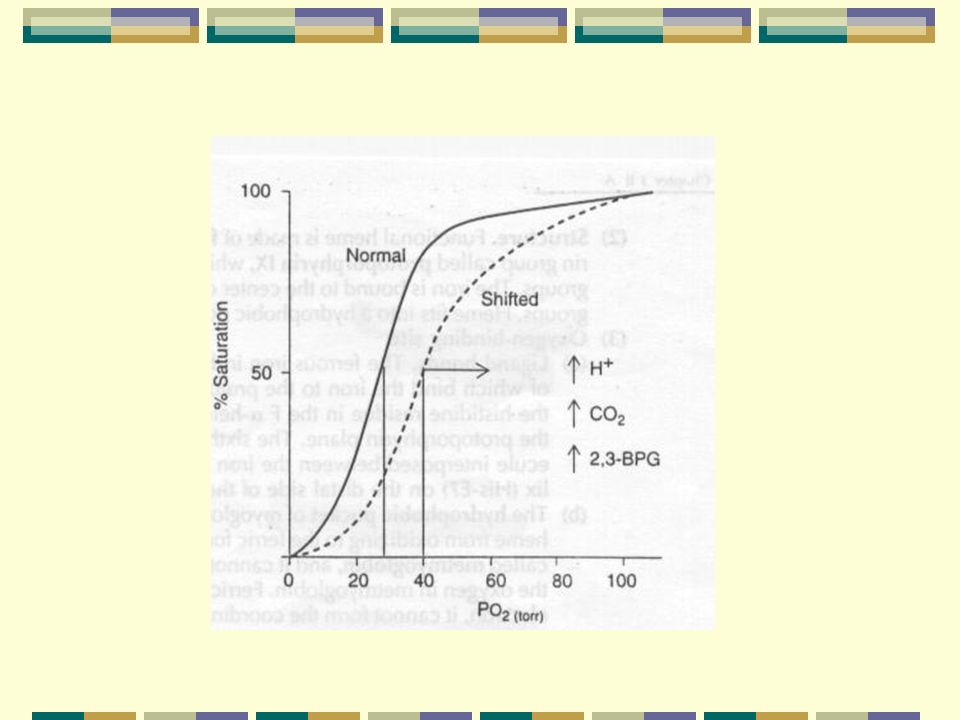
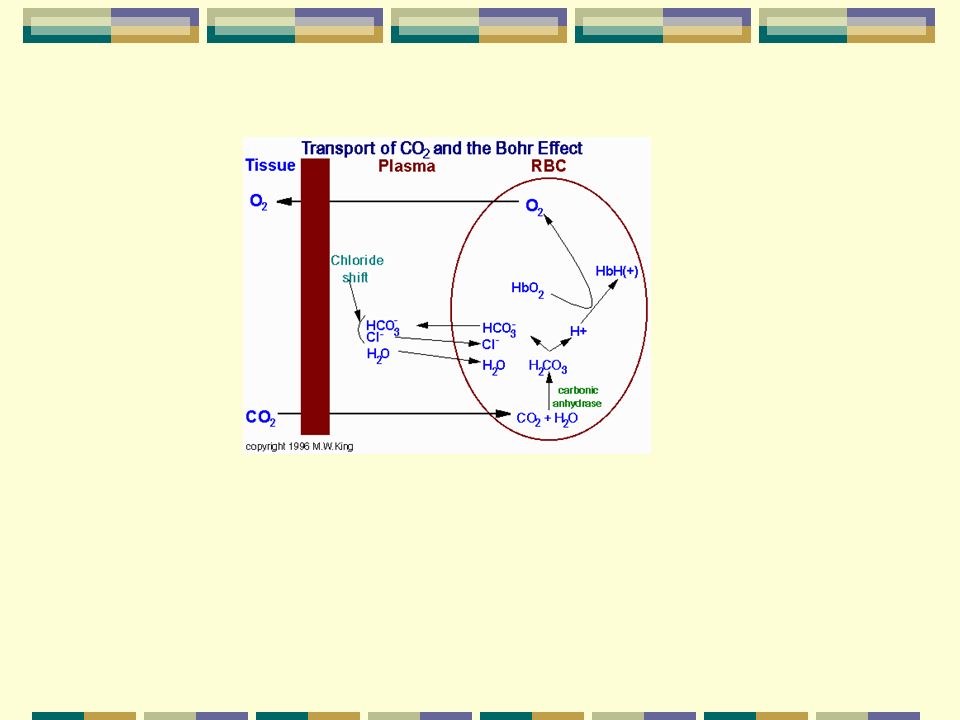
b-Protones H+ y CO2: El efecto Bohr describe la relación
entre la afinidad de la Hb por el O2 a dif. niveles de PCO2 y pH conc. de H+ Afinidad PCO2 (desvío de la curva hacia la derecha) La unión de protones provoca que la Hb pase de la forma R a la T. En los pulmones a alta PO2 y pH 7.4 pasa a la forma R con menor afinidad por los protones y estos son expulsados. La unión de CO2 igualmente favorece el paso a la forma T. CO2 + NH N - COOH +2 H Carbamato
 La unión de protones provoca que la Hb. pase de la forma R a la T. En los pulmones a alta PO2 y pH 7.4 pasa. a la forma R con menor afinidad por los. protones y estos son expulsados. La unión de CO2 igualmente favorece el. paso a la forma T. CO2 + NH3 N - COOH +2 H. Carbamato.")
26
Visión frontal de la moléc
Visión frontal de la moléc. de Hb: en gris se observan los átomos de Carbono, el Nitrógeno en púrpura, y la bola roja es el hierro Imagen lateral con el aminoácido histidina unido al Fe El oxígeno (naranja) se une al Fe y a la histidina
 se une al Fe y a la histidina.")
27
c- Papel del 2,3 BPG: Se encuentra en los GRs en una concentración parecida a la de la Hb Responsable de bajar significativamente la afinidad de la Hb por el O2 y promueve el paso a la forma T Solo una molécula de 2,3 BPG interactúa con cada tetrámero de Hb Aumenta en el GR conforme se adaptan a la hipoxia tisular (anemia, grandes altitudes y disfunción pulmonar) Disminuye en la sangre almacenada por lo que disminuye la capacidad de liberar O2 a los tejidos
 Disminuye en la sangre almacenada por lo que disminuye la capacidad de liberar O2 a los tejidos.")
28
Forma oxigenada y desoxigenada

29
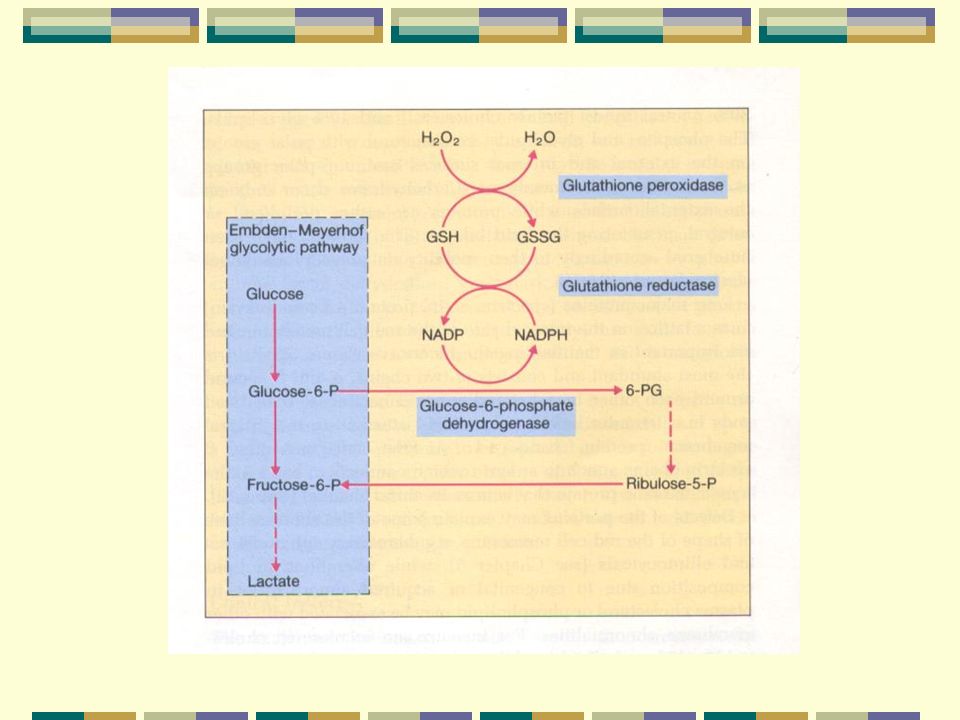
Vía Glicolítica

31
Ciclo del 2-3 DPG

32
2-3 DPG

33
Unión del 2-3 DPG

34
d- Hemoglobina Fetal Alta afinidad por el O2 (subunidades gamma no unen bien el 2-3 DPG) La curva de afinidad por el oxígeno de la HbF está desviada a la izquierda comparada con la de la HbA.

35
Aspectos clínicos Glicosilación HbA en diabetes mellitus.
Hemoglobinopatías: Enfermedades genéticas en las que las subunidades de la Hb han sufrido una mutación. Se han descrito cientos de ellas. Algunas pasan inadvertidas pero otras producen enfermedad desde leve a grave. Tipo de alteración: Por cambios estructurales Por disminución o ausencia en la síntesis de cadenas de globina (Talasemias)
")
36
Hemoglobinopatías más frecuentes (por alteración estructural)
")
37
Hemoglobinopatías

38
Patrones electroforéticos de algunas hemoglobinopatías

41
Ubicación genética de las
Cadenas de globina Herencia de la a talasemia

42
Hemoglobinopatías

43
β α Talasemia Hb adulto: 2 + 2 Definición
Definición Grupo de anemias hemolíticas hereditarias. Disminución de la síntesis de 1 ó + cadenas polipeptídicas de Hb. Cuadro clínico desde indetectables hasta anemia severa y fatal. (Síndromes talasémicos) β α Alfa talasemia: Disminución en la síntesis cadenas α (Exceso cadenas β) Beta talasemia: Disminución en la síntesis de cadenas β (Exceso cadenas α)
 β. α. Alfa talasemia: Disminución en la síntesis cadenas α. (Exceso cadenas β) Beta talasemia: Disminución en la síntesis de cadenas β. (Exceso cadenas α)")
44
Talasemia Causa molecular
Provocada por mutaciones puntuales o deleciones en en 1 o varios de los genes de la α globina y la β globina. La producción de un tipo de proteína Producción de otra proteína constituyente Exceso produce Hb inestable Desnaturalizaciónprecipitación y destrucción GR

45
Talasemia

46
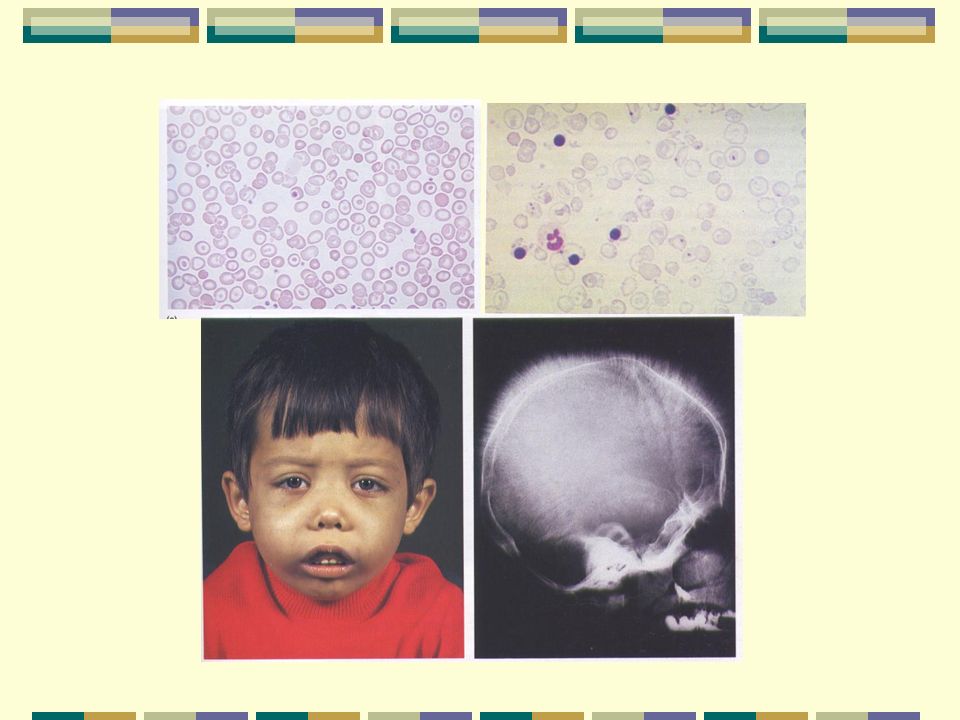
Talasemia mayor- Anemia del Mediterráneo-Anemia de Cooley
1920: Dr. Denton Cooley describió por 1 vez la enfermedad Reconoció signos clínicos (niños italianos y griegos) Anemia Bazo agrandado Deformidades en hueso Antonio Maccanti describía al mismo tiempo esta hemoglobinopatía
 Anemia. Bazo agrandado. Deformidades en hueso. Antonio Maccanti describía al mismo tiempo esta hemoglobinopatía.")
47
Talasemia Mayor Carácter recesivo
Común en Países del Mediterráneo, Sudeste Asiático, India y el Medio Oriente. Costa Rica (De 10 a 12 casos reportados). Último caso hace 2 años. Talasemia Hb S Hb C Hb E Figura 1. Distribución primaria aproximada de talasemia y desórdenes de B-globina en el mundo
. Último caso hace 2 años. Talasemia. Hb S. Hb C. Hb E. Figura 1. Distribución primaria aproximada de talasemia y desórdenes de B-globina en el mundo.")
48
Talasemia Mayor (Derivado de palabra griega para mar “thalassa” )
Características más importantes: Anemia severa Hb de 4 – 5 g/dL Dx entre los 6 – 12 meses de edad Hepato esplenomegalia Eritropoyesis ineficaz Dependientes vitalicios de transfusiones (daño de órganos) Patrón de electroforesis de Hb: FA2, FAA2 A F A2 + -
 Patrón de electroforesis de Hb: FA2, FAA2. A F A")
51
MUCHAS GRACIAS

Presentaciones similares