Descargar la presentación
La descarga está en progreso. Por favor, espere

1
β TALASEMIAS

2
Definición Es un trastorno autosómico recesivo de la sangre caracterizado por una síntesis anómala de las cadenas β globina de la hemoglobina. Thalassa: mar Haima: sangre

3
Forma de Herencia

4
Formas de Beta Talasemia
Talasemia Mayor, de forma variable, Anemia de Cooley o Anemia del Mediterraneo. Talasemia Intermedia Talasemia Leve Beta Talasemia asociada a otras hemoglobinas anómalas: HbC, HbE, HbS y persistencia hereditaria de HbF.

5
Epidemiología Mayor incidencia en: países Mediterraneos, Oriente Medio, Asia central, India, Sur de China y el lejano Oriente, países en la costa norte de Africa y en América del sur.

6
Epidemiología La mayor frecuencia de portadores del gen de la beta talasemia en estas regiones está relacionado a la presión selectiva de Plasmodium falciparum. Cerca del 1,5 % de la población mundial (es decir de 80 a 90 millones de personas) son portadores de BT, con cerca de pacientes sintomáticos nacidos al año, la gran mayoría en países en vías de desarrollo.
 son portadores de BT, con cerca de pacientes sintomáticos nacidos al año, la gran mayoría en países en vías de desarrollo.")
7
Descripción Clínica β Talasemia Mayor:
La presentación clínica ocurre entre el 6to mes y el 2° año de vida y se caracteriza por: Anemia, retraso en el crecimiento, palidez creciente, problemas en la alimentación, diarrea, irritabilidad, recurrentes episodios de fiebre, y distención abdominal creciente causada por inflamación hepática y esplénica,

8
Tratamiento talasemia mayor
TRANSFUSIONES: Lo que se busca es la corrección de la anemia, supresión de la eritropoyesis y la inhibición de la absorción de hierro gastrointestinal mediante dietas específicas bajas en hierro. El comienzo del tratamiento de debe evaluar según la condición de cada paciente. Evita el deterioro del crecimiento, daño en órganos y deformidades de los huesos, lo que permite mejorar la calidad de vida. Frecuencia: cada 2 a 4 semanas

9
Descripción Clínica Las transfusiones sanguíneas regulares, trae como consecuencia la elevación en los niveles de hierro, generando retardo en el crecimiento y maduración sexual, problemas cardíacos como miocardiopatía dilatada, trastornos hepáticos, esplénicos, y en las glándulas endocrinas. Las complicaciones cardíacas debidas a sobrecarga de hierro constituyen la causa de muerte en el 71 % de los pacientes de β talasémicos.

11
Tratamiento Para contrarrestar la sobrecarga de hierro se recurre a quelantes que permiten la excreción de hierro a través de la orina y/o las heces El tratamiento con quelantes se debe iniciar luego de las 10 a 20 transfusiones o cuando aumentan los niveles de ferritina por encima de los 1000ng/ml. También se deben tratar los trastornos ocasionados por el hierro en exceso.

12
Genu Valgum

13
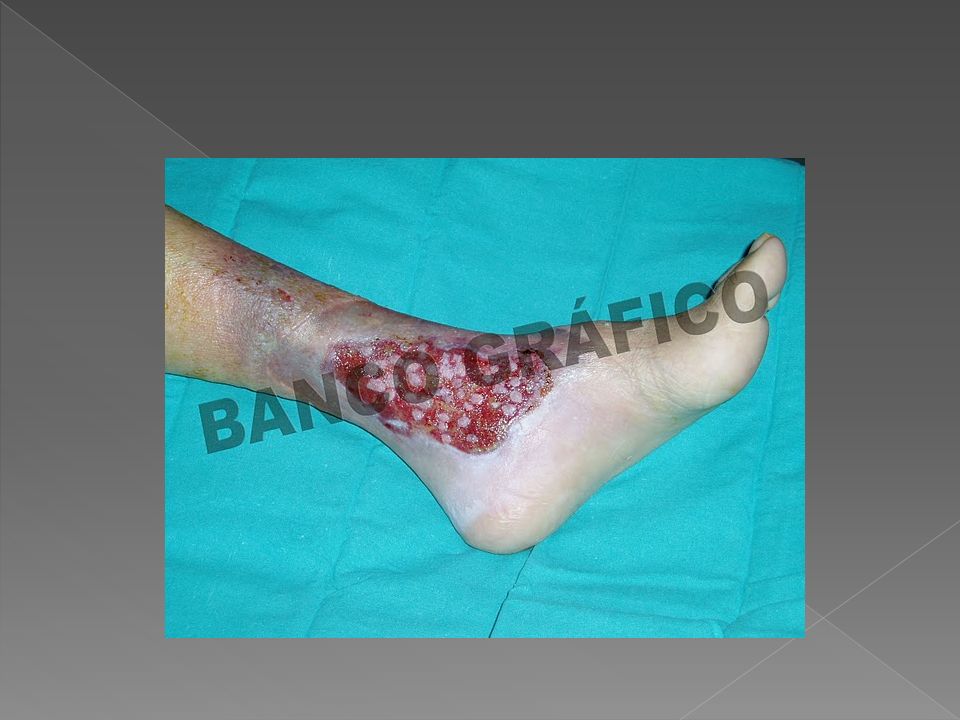
Descripción Clínica β talasemia intermedia:
Se presenta más tardíamente y de forma variable. Hay casos en que la enfermedad se manifiesta de forma severa antes de los seis años, y otros en los que cursan hasta la vida adulta en forma asintomática solamente con una leve anemia. En algunos pacientes hay deformidades óseas debido a que la eritropoyesis está muy aumentada, habiendo incluso eritropoyesis extramedulares y osteoporosis. Hay complicaciones en bazo, hígado y ganglios linfáticos, úlceras en los pies, cálculos biliares, predisposición a trombosis.

14
¿Por que se expande la médula ósea?
Como consecuencia de la ausencia o deficiencia de las cadenas β de hemoglobina, las cadenas alfa están en exceso y precipitan. Esto conduce a una hemólisis periférica, entonces se genera anemia. El organismo responde produciendo eritropoyetina, la cual produce una aumento en el tamaño de la médula ósea.

16
Descripción Clínica β talasemia menor:
Generalmente se presenta en forma asintomática o bien se presenta en una anemia leve.

17
β talasemia asociada a otros transtornos:
En raras ocasiones la beta talasemia no se remite únicamente al gen de la beta hemoglobina: Rasgo de Beta talasemia asociado a tricotiodistrofia: se encuentra en el gen de la transcripción TFIIH . Trompocitopenia con talasemia ligada al X: se ve afectado el factor de transcripción ligado al X GATA-1

18
Etiología Se han descripto mas de 200 mutaciones, la mayoria son puntuales en la region funcional del gen de la globina. Las delecciones son poco comunes.

19
Tipos de mutación de acuerdo al grupo étnico
NOTACIÓN: β° = ausencia total de la síntesis de beta globina por el alelo afectado. β+ = producción residual de la cadena de beta globina por el alelo mutado, alrededor del 10% β ++ = reducción muy baja en la producción de beta globina

20
Modificadores genéticos
Son variantes genéticas que dan lugar a diferencias en el fenotipo de la enfermedad. Existen variaciones que reducen el desequilibrio entre las cadenas, resultando una talasemia más leve: Como es la presencia de HbF en la vida adulta. También puede darse una disminución en la síntesis de cadenas alfa, por lo que es menor la precipitación de las mismas.

21
Diagnostico: Hematológico: Recuento de GR indican anemia
Hemoglobina: menor a 7 g/dl indica talasemia mayor entre 7 y 10 g/ dl indica talasemia intermedia Determinación del volumen corpuscular medio(VCM); ancho de distribución de los hematíes(RDW); determinación de velocidad de sedimentación globular(VCG).
; ancho de distribución de los hematíes(RDW); determinación de velocidad de sedimentación globular(VCG).")
22
Frotis sanguíneo: Presencia de glóbulos rojos amorfos. Determinación cuali y cuantitativa de hemoglobina mediante electroforesis, HPLC,y niveles de hemoglobina fetal por desnaturalización alcalina. Electroforesis de Hb en cellogel, seguido de cuantificación de los niveles de HbA2

23
Análisis molecular Mediante PCR, Dot blot, y por amplificación primer específica. Al ser mutaciones puntuales no se detectan con Southern Blot a diferencia de la talasemia alfa. Si el análisis de la mutación puntual falla, se recurre a la secuenciación del gen para detectar la mutación específica.

24
Asesoramiento genético y diagnóstico prenatal
El diagnóstico pre natal es posible mediante el análisis de ADN extraído de células fetales mediante amniocentésis entre la semana 15 y 18, o bien, de las vellosidades coriónicas a la semana 11.

25
FIN

Presentaciones similares





>")







. Forma primaria: herencia autosómica recesiva Alteración.>")






