Descargar la presentación
La descarga está en progreso. Por favor, espere

1
Dr. Raúl Ramos Vázquez Septiembre 2006 HOSPITAL UNIVERSITARIO
Departamento de Patología Clínica ANEMIAS HEMOLÍTICAS Dr. Raúl Ramos Vázquez Septiembre 2006

3
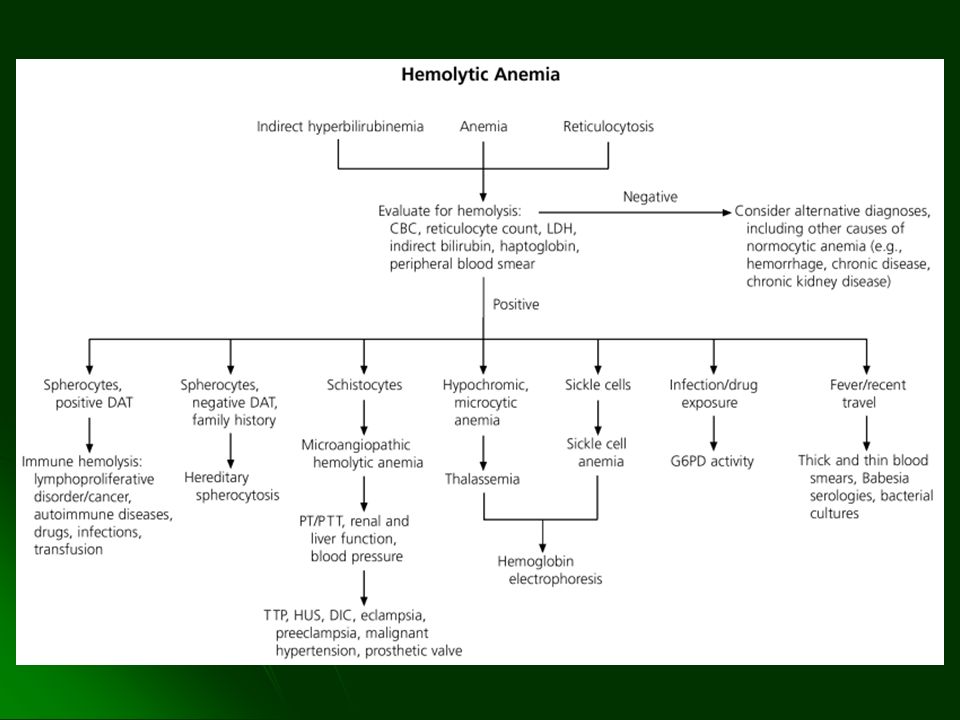
HEMOLISIS Destrucción o remoción de eritrocitos de la circulación antes de completar su periodo de vida. Generalmente se acompaña de anemia cuando la eritropoyesis no puede compensarla

4
HISTORIAL CLINICO Datos de síndrome anémico Historia familiar
Ictericia Hemoglobinuria Linfadenopatía Esplenomegalia Ulceras en extremidades

5
Extravascular Intravascular
EVALUACION DE LABORATORIO DE HEMOLISIS Extravascular Intravascular HEMATOLOGICO Frotis de SP Cuenta de reticulocitos Exámen de médula ósea Policromatofilia Hiperplasia eritroide SERICO Bilirrubina Haptoglobina Hemoglobina libre plasma LDH la Indirecta N ó N ó (variable) ó Ausente (variable) URINARIO Hemosiderina Hemoglobina + + (en casos severos)
 ó Ausente. (variable) URINARIO. Hemosiderina. Hemoglobina. + + (en casos severos)")
6
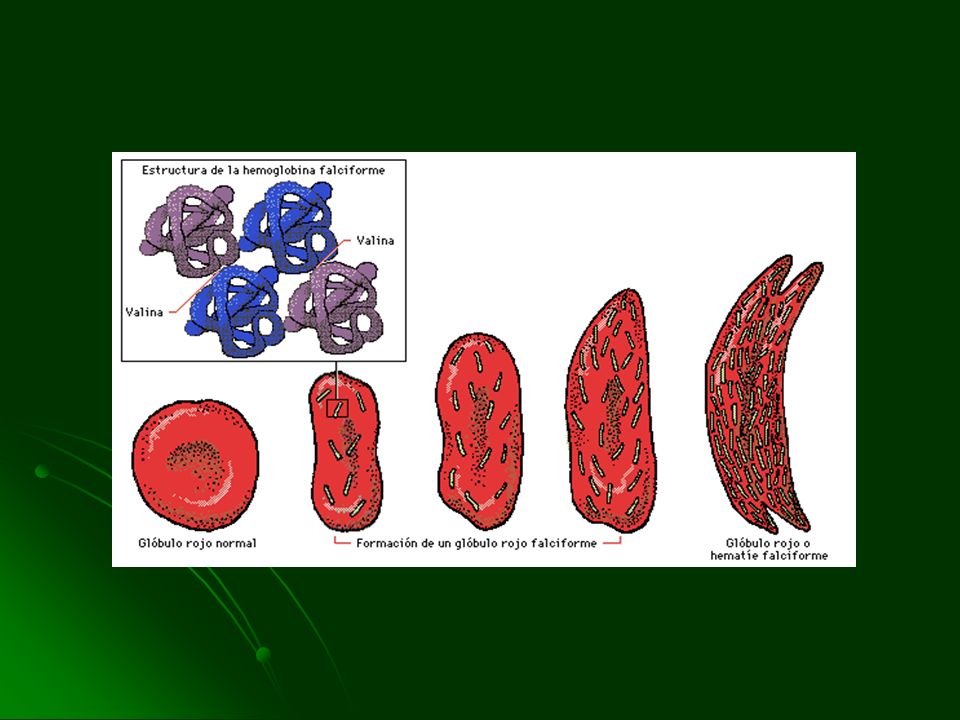
DREPANOCITOSIS

7
6. HEMOGLOBINURIA PAROXISTICA NOCTURNA

8
Red Blood Cell Morphology in the Diagnosis of Hemolytic Anemia
Cause Syndromes Spherocytes Target cells Schistocytes Sickled cells Acanthocytes Agglutinated cells Heinz bodies Loss of membrane Increased ratio of RBC surface area to volume Traumatic disruption of membrane Polymerization of hemoglobin S Abnormal membrane lipids Presence of IgM antibody Precipitated hemoglobin Hereditary spherocytosis, immunohemolytic anemia Hemoglobin disorders: thalassemias; liver disease Microangiopathy, intravascular prostheses Sickle cell syndromes Severe liver disease (spur cell anemia) Cold agglutinin disease Unstable hemoglobin, oxidant stress
 Cold agglutinin disease. Unstable hemoglobin, oxidant stress.")
9
ANEMIAS HEMOLITICAS HEREDITARIAS
Alteración en membrana En enzimas En hemoglobina DEFECTOS DE MEMBRANA Esferocitosis Eliptocitosis Estomatocitosis

10
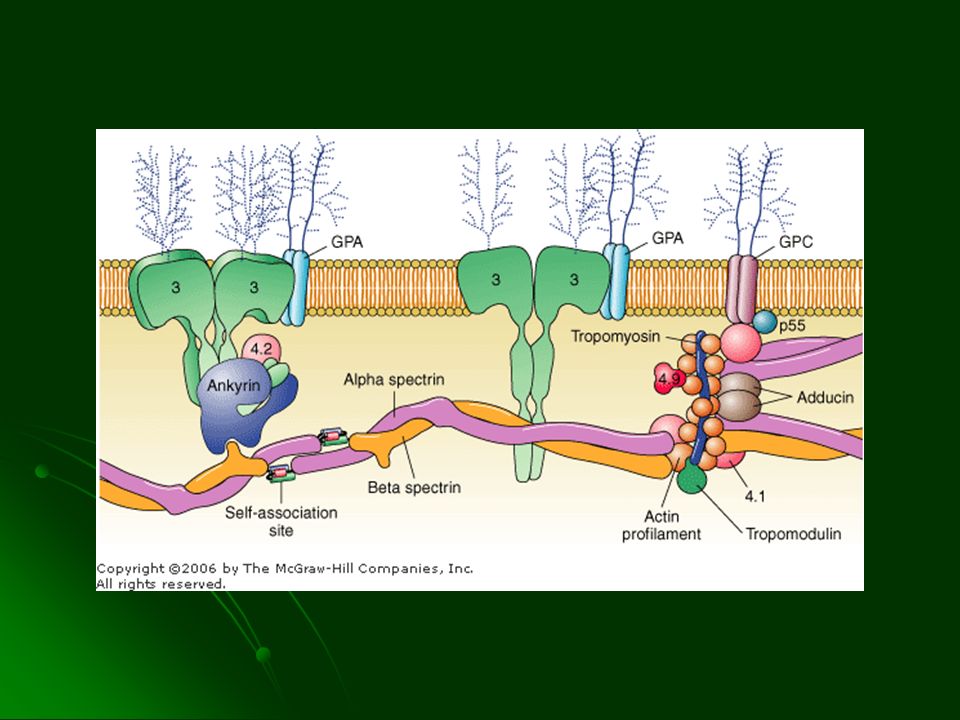
ESFEROCITOSIS Reducción del área superficial contra el volumen
Esférico y no deformable Rigidez captación por el bazo Autosomico dominante 20% autosómico recesivo 1:1000 – 1:4500 50% Defecto en ankirina

14
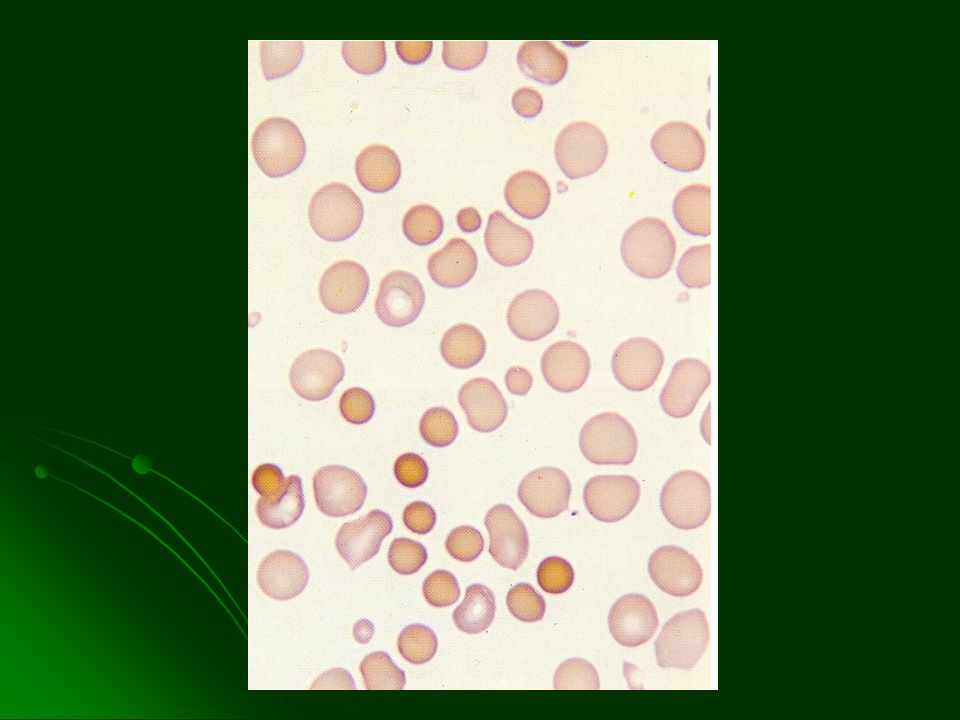
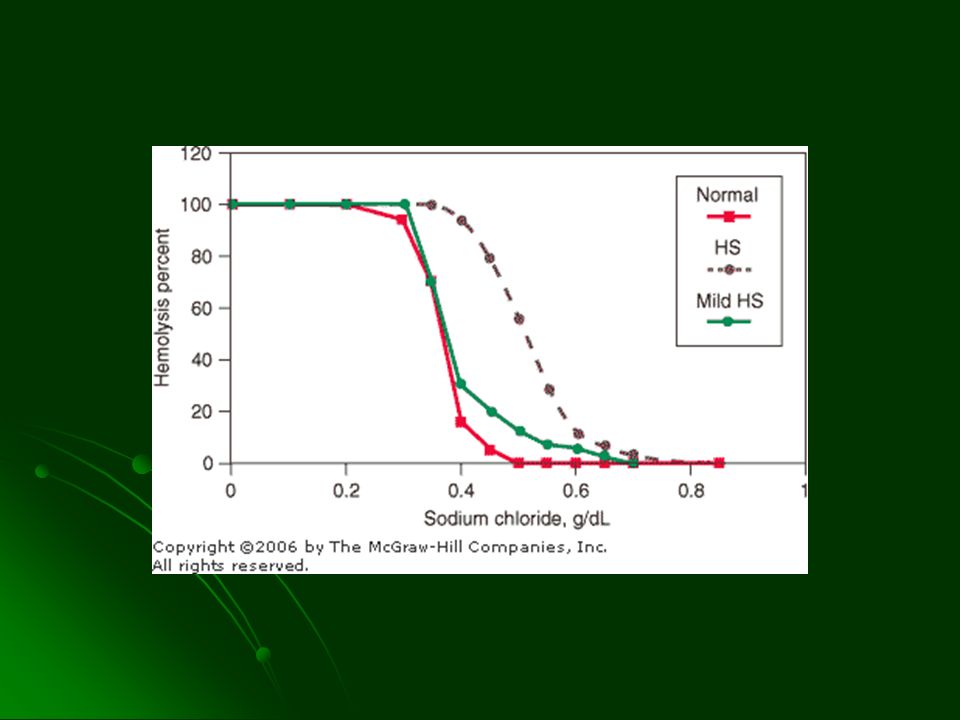
DIAGNOSTICO Esferocitos VCM normal o y CMHC 35 – 40% Prueba de la fragilidad osmótica del eritrocito Historia familiar Distinguir de hemólisis inmune con test de Coombs directo

16
TRATAMIENTO Esplenectomía en pacientes sintomáticos Niños después de 4 años Evitar organismos encapsulados Pneumococcus, Menigococcus, Haemophilus influenzae Vacunación 2 semanas antes de la Qx

17
ANEMIAS HEMOLITICAS HEREDITARIAS
DEFECTOS ENZIMATICOS Deficiencia de G6PD Deficiencia de Piruvato Cinasa Hexoquinasa

19
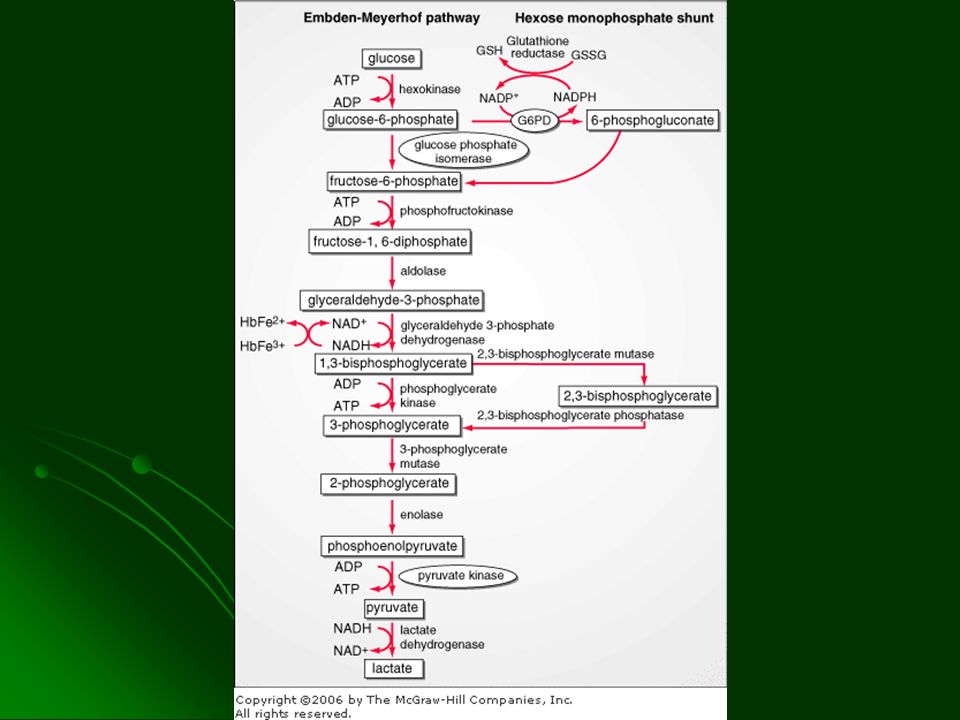
DEFICIENCIA DE G6PD El glóbulo rojo normal al ser expuesto a drogas o toxinas que liberan radicales libres aumenta la actividad de la vía de las pentosas Se genera glutation reducido Protección de la membrana del GR y de los grupos sufhidrilo de la Hb contra la oxidación Se evita la precipitación de la Hb

20
Más de 200 millones de casos a nivel mundial
Protege parcialmente contra la malaria Hay 400 variantes de G6PD Herencia ligada a X Se acorta la vida del GR Las manifestaciones clínicas aparecen ante condiciones de estrés para el GR

21
EPISODIOS HEMOLITICOS DEBIDOS A:
Infecciones virales o bacterianas Antimaláricos (primaquina) Sulfonamidas Nitrofurantoína Miscelaneos (azul de metileno, ácido nalidíxico, fenazopiridina) Favismo (habas)
 Sulfonamidas. Nitrofurantoína. Miscelaneos (azul de metileno, ácido nalidíxico, fenazopiridina) Favismo (habas)")
22
MANIFESTACIONES Crisis hemolítica aguda horas después de la exposición (hemoglobinuria) Autoimitación de la crisis destrucción de GR viejos En negros con variante A- la masa de GR se reduce 25 a 30% Oxidación de la Hb cuerpos de Heinz El tipo mediterráneo es más propenso al favismo DX: Medición de actividad enzimática e Interrogatorio

24
TRATAMIENTO Autolimitación de la crisis No Tx específico Esplenectomía no beneficia en casos graves Transfusiones raramente se indican Prevenir el episodio hemolítico Tratamiento de infección subyacente

25
ANEMIAS HEMOLITICAS HEREDITARIAS
HEMOGLOBINOPATIAS Drepanocitosis o Anemia de células falciformes Hemoglobinopatía C, E, etc DEFECTO EN LA SINTESIS DE GLOBINAS Talasemias

26
DREPANOCITOSIS Autosómico codominante Heterocigotos (AS) portadores
Homocigotos (SS) enfermos Africa tropical prevalencia de portadores de 20 – 40% México prevalencia en costas del Golfo Otorga resistencia ante plasmodium falciparum
 enfermos. Africa tropical prevalencia de portadores de 20 – 40% México prevalencia en costas del Golfo. Otorga resistencia ante plasmodium falciparum.")
27
FISIOPATOLOGIA Mutación puntual, sustitución de valina por ácido glutámico en la cadena de Hb Cuando la HbS esta bien oxigenada es soluble Al disminuir la PO2 se polimeriza y forma tactoides o cristales Rigidez del GR Impedimento a la circulación estancamiento Si dura corto tiempo es reversible

29
MANIFESTACIONES La anemia inicia en los primeros meses de vida Hepatoesplenomegalia infartos de bazo autoesplenectomía Raza negra, crecimiento y maduración sexual retardados Ictericia conjuntival, úlceras maleolares Crisis vasculares oclusivas infarto

30
CRISIS VASCULARES Inician con Fiebre y dolor intenso Antecedente de hipoxia excesiva: Procesos infecciosos Deshidratación Ejercicio excesivo Trabajo de parto Grandes altitudes

31
Niños de 9 m dactilitis, Sx mano- pie
Infancia Infecciones frecuentes por St pneumoniae Osteomielitis por Salmonella Adultos Infartos pulmonares Depósito de hierro en miocardio por múltiples transfusiones El Dx de la crisis vascular depende del órgano involucrado Crisis aplásicas parvovirus B19

32
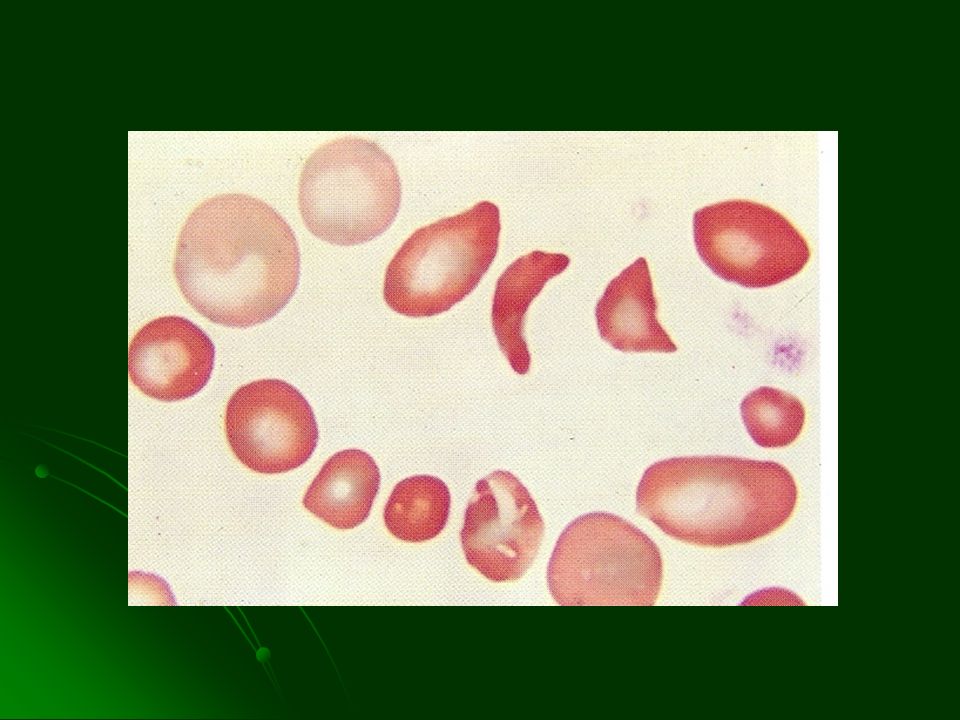
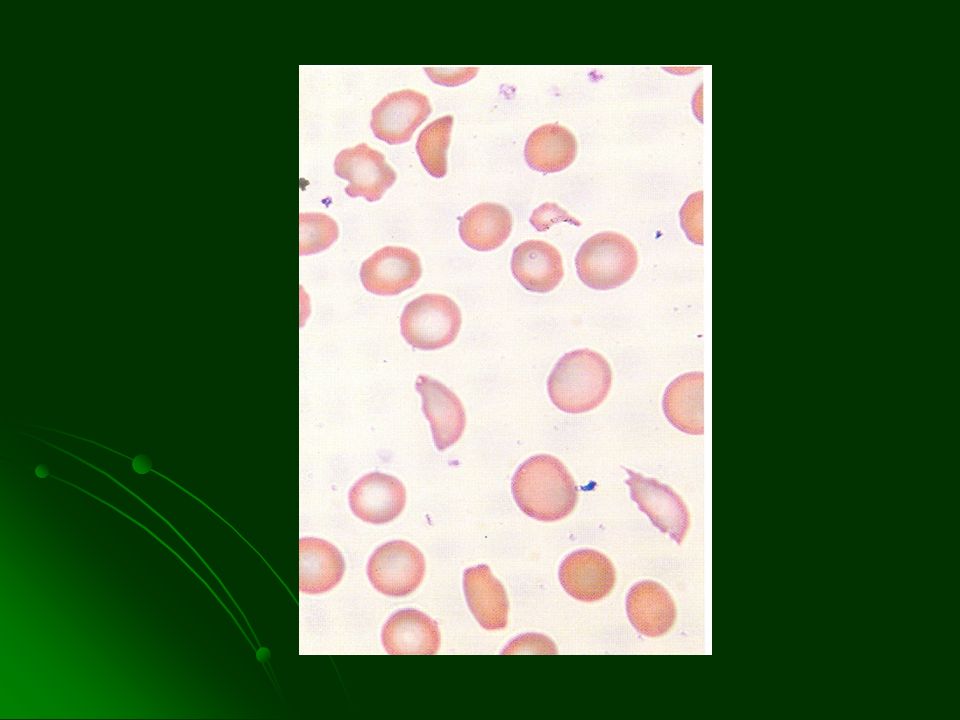
LABORATORIO Anemia normocítica normocrómica inicia entre los 6 y 9 m de edad Anisocitosis y drepanocitos Policromatofilia, CRN, reticulocitosis Hiperbilirrubinemia indirecta LDH Prueba de inducción de drepanocitos + Prueba de solubilidad reducida + Estudio electroforético de la HbS

34
TRATAMIENTO Medidas profilácticas Prevención de infecciones y vacunación Reposo, hidratación, oxigenación, analgésico Transfusión sanguínea

35
TALASEMIAS Grupo de anormalidades de la Hb, hereditarias
Producción insuficiente de cadenas de globina Se clasifican con el nombre de la cadena cuya síntesis está reducida α, β, δ, δ/β, γ/δ/β Mayor y menor indican gravedad pero no estado hetero u homocigoto 3% de la población mundial es portador (Mediterráneo)
")
36
TALASEMIA La de más frecuencia Se ve en México Deficiencia en producción de cadenas por reducción de su síntesis Elevado polimorfismo genético La expresión clínica varía Se Dx determinando la fracción A2 y F de la Hb Homocigota = Anemia de Cooley

37
MANIFESTACIONES Y Dx Esplenomegalia Ensanchamiento del diploe y estrías verticales (cráneo en cepillo) Exceso de Hierro secundario a transfusiones Anemia microcítica hipocrómica Dianocitos Electroforésis Incremento de Hb F PCR

39
TRATAMIENTO Transfusiones de PG Medicamentos quelantes de hierro Distinguir de Anemia Ferropénica Ferrodinamia normal en talasemia RDW normal

40
ANEMIAS HEMOLITICAS ADQUIRIDAS
CAUSA INMUNE Reactividad A Ac calientes (IgG) Reactividad a Ac fríos (IgM) Reactividad a Ac fríos IgG (Hemoglobinuria paroxística a frigori) Dependiente de fármacos (hapteno, autoinmune) CAUSAS MECANICAS Hemoglobinuria de la marcha Prótesis Microangiopátcas (PTT, SUH, CID) Efecto tóxico en membrana (BIOLOGICOS, QUIMICOS) HEMOGLOBINURIA PAROXISTICA NOCTURNA
 Reactividad a Ac fríos (IgM) Reactividad a Ac fríos IgG (Hemoglobinuria paroxística a frigori) Dependiente de fármacos (hapteno, autoinmune) CAUSAS MECANICAS. Hemoglobinuria de la marcha. Prótesis. Microangiopátcas (PTT, SUH, CID) Efecto tóxico en membrana (BIOLOGICOS, QUIMICOS) HEMOGLOBINURIA PAROXISTICA NOCTURNA.")
41
ANEMIA HEMOLITICA AUTOINMUNE
Primario o secundario Demostrar anticuerpos sobre la membrana del GR Prueba de antiglobulina directa (Coombs), ELISA y CF 90 – 95% son IgG 10% IgM (aglutininas frías) Acrocianosis, Hburia
, ELISA y CF. 90 – 95% son IgG. 10% IgM (aglutininas frías) Acrocianosis, Hburia.")
42
MANIFESTACIONES 50% relacionado a padecimientos autoinmunes Se presenta en algún momento en más de 40% de pacientes con LES 10 – 15% asociado a medicamentos (metildopa, l-dopa, procainamida) IgM asociado a Epstein Barr o Mycoplasma pneumoniae
 IgM asociado a Epstein Barr o Mycoplasma pneumoniae.")
43
FISIOPATOLOGIA Hemólisis extravascular en IgG (SFM esplénico) En IgM es hemólisis intravascular Predisposición genética Desequilibrio en la actividad de linfocitos T (Th1 y Th2)
")
44
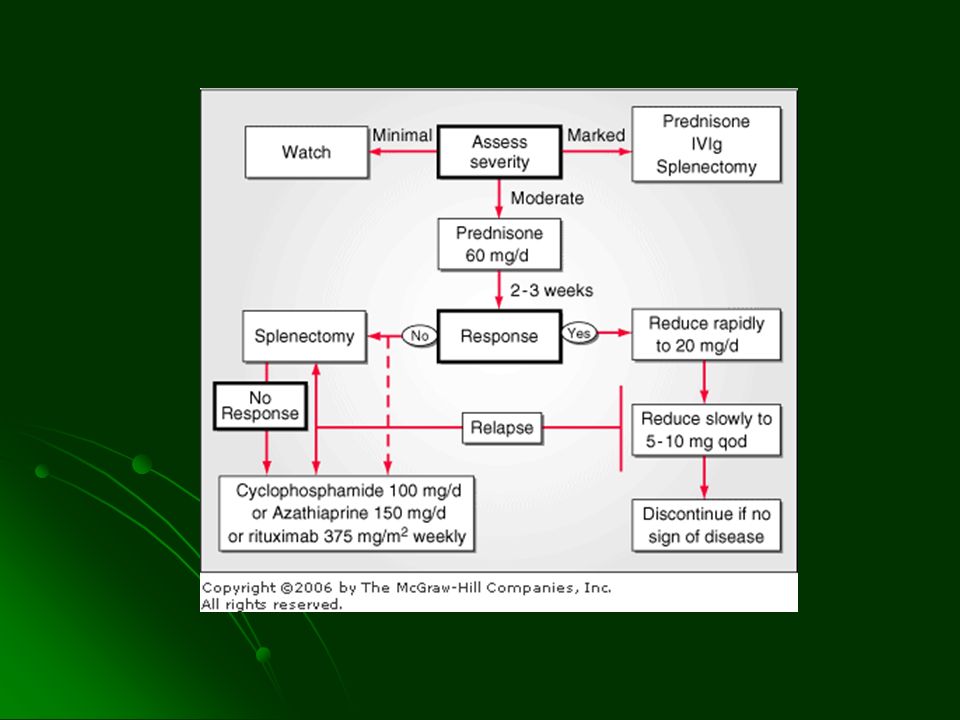
TRATAMIENTO Corticosteroides (Prednisona) Reducción en producción de Ac Disminución de la unión del Ac a la membrana Menor captación de GR sensibilizados por el SFM Esplenectomía Azatioprina, ciclofosfamida citotóxicos (eritropoyesis) Evitar transfusiones de PG hemólisis
 Evitar transfusiones de PG hemólisis.")
46
HEMOGLOBINURIA PAROXISTICA NOCTURNA
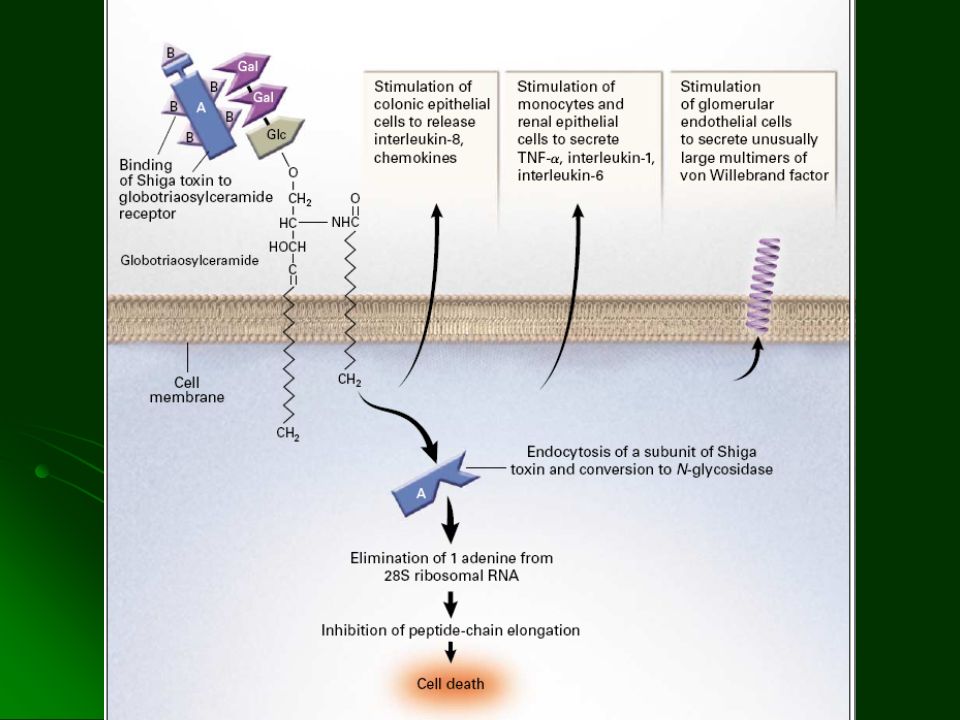
AH por defecto intracorpuscular pero Adquirida 2 casos por 100,000 habitantes (México) Padecimiento clonal del tejido hematopoyético Mayor sensibilidad al efecto citolítico del complemento Hemólisis intravascular Hematopoyesis ineficaz
 Padecimiento clonal del tejido hematopoyético. Mayor sensibilidad al efecto citolítico del complemento. Hemólisis intravascular. Hematopoyesis ineficaz.")
47
PATOGENESIS Mutación del gen PIG-A del brazo corto del cromosoma X Alteración en síntesis de moléculas de GPI (glucosilfosfatidilinositol) Deficiencia de proteínas de membrana que se fijan al GPI Ocurre después de un daño medular o como mecanismo defensivo

48
Deficiencia de proteínas que regulan la actividad del complemento y no pueden fijarse al GPI
CD55 o DAF (acelera) y CD59 o MIRL (restrictor) Mayor fijación del C al Gr Deficiencia de CD 59 en las plaquetas hipercoagulabilidad y trombosis
 y CD59 o MIRL (restrictor) Mayor fijación del C al Gr. Deficiencia de CD 59 en las plaquetas hipercoagulabilidad y trombosis.")
49
MANIFESTACIONES Anemia hemolítica, trombosis venosa, hematopoyesis ineficaz Hemoglobinuria intermitente, Hemosiderinuria presente Se manifiesta cuando hay activación del complemento (EJ: Infección) Leucopenia y trombocitopenia Trombosis en 40% europeos y menos en asiáticos Venas intraabdominales (Sx de Budd Chiari) Esplenomegalia congestiva También en senos venos cerebrales Periodos de aplasia medular y asociación con Sx mielodisplásico
 Leucopenia y trombocitopenia. Trombosis en 40% europeos y menos en asiáticos. Venas intraabdominales (Sx de Budd Chiari) Esplenomegalia congestiva. También en senos venos cerebrales. Periodos de aplasia medular y asociación con Sx mielodisplásico.")
50
DIAGNOSTICO Identificación de GR sensibles al C en sistemas de hemólisis in vitro Hemólisis ácida o de Ham Hemólisis de sucrosa Hemosiderina en sedimento urinario Citometría de flujo identificación de células carentes de CD 55 y CD 59

51
TRATAMIENTO Anabólicos orales (mesterolona, danazol) Prednisona Hierro oral para deficiencia de Fe La crisis hemolítica responde adecuadamente a la transfusión (↓eritropoyesis y los nuevos GR controlan el C) Tx del fenómeno trombótico Transplante de Médula Ósea
 Tx del fenómeno trombótico. Transplante de Médula Ósea.")
52
MICROANGIOPATIAS PURPURA TROMBOCITOPENICA TROMBOTICA
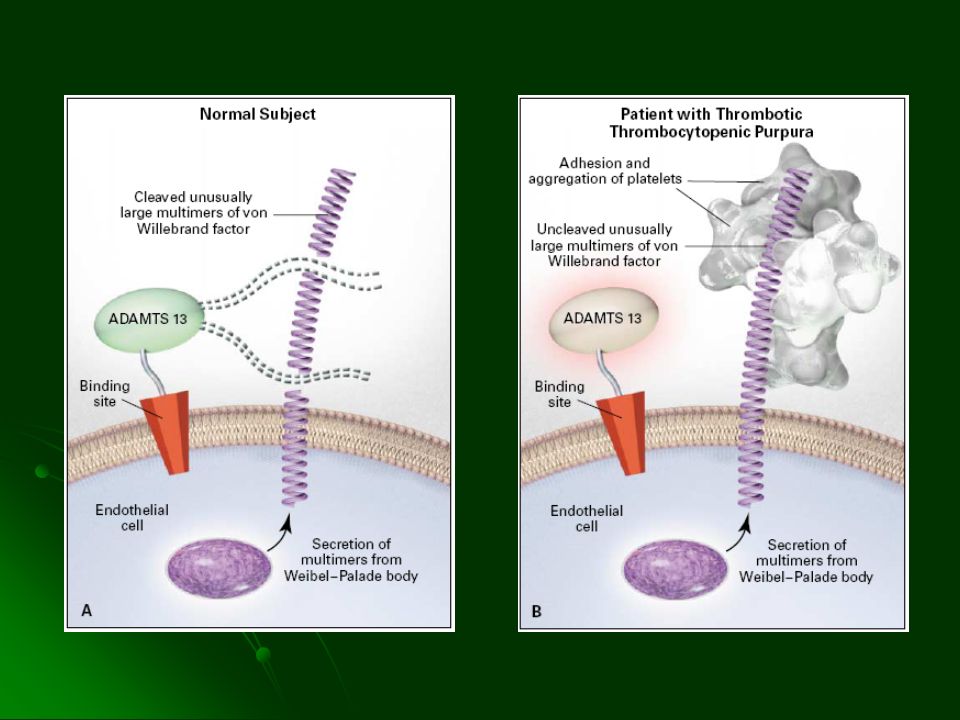
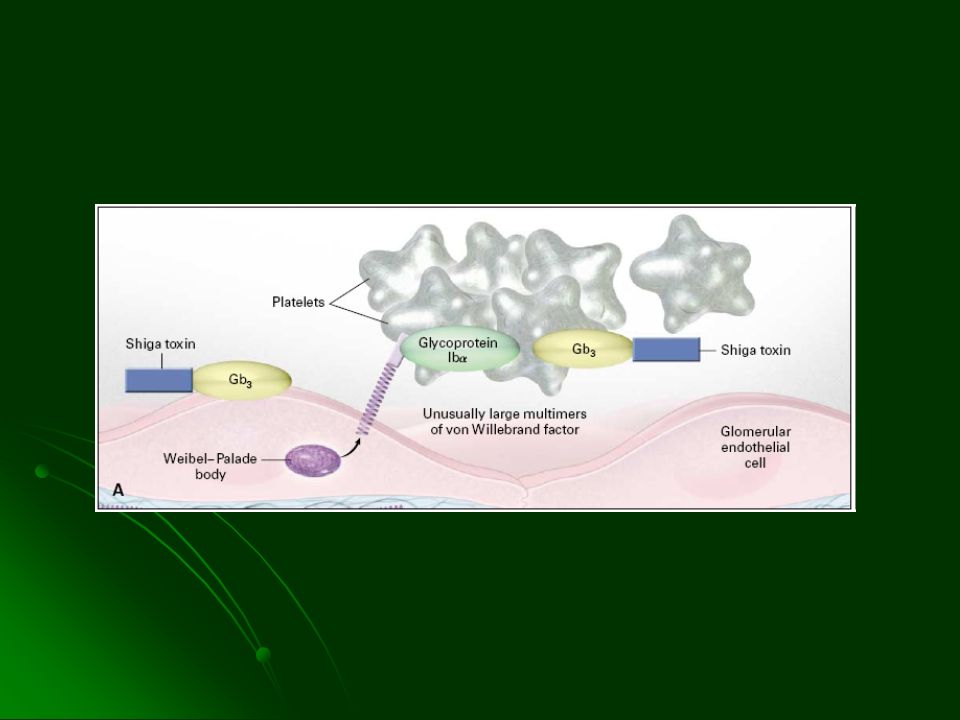
Anemia Hemolítica microangiopática Trombocitopenia Trastornos neurológicos Fiebre Alteraciones renales Trombos hialinos con plaquetas, escasa fibrina y grandes cantidades de FvW en circulación Mujeres en la 3° década de la vida

59
TRATAMIENTO Plasmaféresis Desprovisto de fibrinógeno y FvW Supervivencia mayor de 80% Esteroides, Inmunosupresores Esplenectomía

Presentaciones similares












>")











