Descargar la presentación
La descarga está en progreso. Por favor, espere

1
Anemias Hemolíticas

2
Síndrome hemolítico Son todas aquellas situaciones en las que el síndrome anémico se debe a una destrucción anormal de los eritrocitos

3
Clínica del síndrome hemolítico
Anemia Ictericia Cálculos biliares Esplenomegalia( por captación y por compensar mediante la eritropóyesis) Alteraciones óseas Insuficiencia y cálculos renales Ulceras en las piernas
 Alteraciones óseas. Insuficiencia y cálculos renales. Ulceras en las piernas.")
4
Clasificación de las anemias hemolíticas
Según su herencia en: congénitas y adquiridas Según el mecanismo de la lesión: intrínsecas y extrínsecas ( dentro o fuera del hematie) Según el lugar de producción de la hemolisis: intravascular o extravascular
 Según el lugar de producción de la hemolisis: intravascular o extravascular.")
5
Grupos de anemias hemolíticas hereditarias
Por alteraciones en la membrana eritrocitaria Por alteraciones en el metabolismo del hematie Por defecto en la hemoglobina

6
Anemias hemolíticas congénitas
Por defecto en la membrana: esferocitosis hereditaria, eliptocitosis hereditaria por defecto del metabolismo: por déficit de piruvato cinasa, por déficit de G6PDH por defecto de la Hb: hemoglobinopatías y Talasemias

7
Anemias hemolíticas adquiridas
Anemias hemolíticas inmunes Hiperesplenismo Microangiopatías Hemoglobinuria paroxística nocturna.

8
Anemias Hemolíticas Intravasculares: Reacción transfusional aguda
Déficit de G6PDH Extravasculares: Reacción transfusional tardia Hiperesplenismo

9
Hemolisis extravascular
Es un incremento exagerado en la fagocitosis de hematíes por el Sist. Mononuclear fagocítico en bazo e hígado.

10
Protoporfirina Biliverdina Bilirrubina Ind Conjugación Ac. glucurónico Bilirrubina dir Urobilinógeno

11
Hemoglobina Haptoglobina Hemoglobina libre Riñón Lesión tubular

12
Fisiopatología de la hemólisis
Disminución Hb Aumento de Degradación Bilis y DHL Aumento de Producción reticulocitos

16
En la Historia clínica Se debe buscar:
historia familiar de anemia o ictericia ingesta de fármacos o determinados alimentos (p.e habas) infecciones previas raza del individuo (p.e negra déficit de G6PDH y la falciforme)
 infecciones previas. raza del individuo (p.e negra déficit de G6PDH y la falciforme)")
17
Membrana eritrocitaria
Función: regula el volumen y la deformabilidad del hematie. Una alteración de los lípidos de esta dan poca o nula sintomatologia , pero hay presencia de formas raras de eritrocitos, p.e acantocitos.

18
Anemias por alteración de la membrana
La membrana tiene diversos componentes que son: Fosfolípidos, colesterol, glicolípidos, glicoproteínas, enzimas y proteínas del esqueleto, la alteración de algunas proteínas del citoesqueleto dan lugar a esferocitosis y a eliptocitosis hereditarias.

19
Esferocitosis hereditarias
Existe una alteración de las proteínas plasmáticas del citoesqueleto, con perdida de parte de la memb eritrocitaria y aumento en la permeabilidad ionica. Disminuye la relación superficie/volumen del hematie Esto da lugar a fragilidad osmótica y disminución de la elasticidad, los hematíes toman aspecto de globo que al pasar por el bazo son destruidos.

20
Clínica de la esferocitosis
Clínica del síndrome hemolítico Posible esplenomegalia gigante Suele ser anemia ligera VCM suele estar ligeramente disminuido (menos de 80 fL) Microcitosis
 Microcitosis.")
21
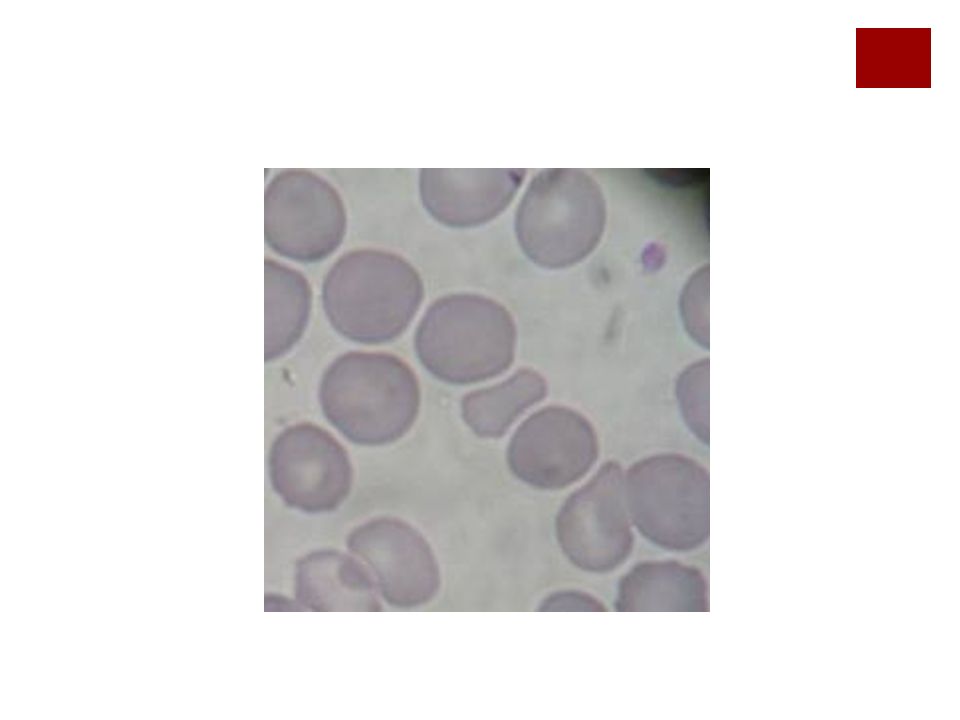
Micro esferocito

22
Pruebas confirmatorias
Morfología de los eritocitos test de fragilidad osmótica Otras pruebas: test de autohemolisis, lisis en glicerol acidificado.

23
Test de fragilidad osmótica

24
Tratamiento de la esferocitosis
Esplenectomía, se tratan solo casos con manifestaciones intensas suele realizarse colecistectomía para evitar cálculos biliares Acido fólico para la eritropoyesis

25
Eliptocitosis hereditaria
Se produce por una alteración en una proteína de membrana: la espectrina no puede formar tetrameros, y el eritrocito no puede recuperar su forma tras una deformación, por lo que adquiere forma de elipse.

26
Solo produce manifestaciones clínicas en el 10% de los pacientes.
Tx ac fólico En casos con clínica intensa puede hacerse esplenectomía

27
Eliptocitos

28
Anemias por alteración del metabolismo
Metabolismo del eritrocito Shunt de Pentosas Glicólisis

29
Deficiencia G6PD Deficiencia Piruvato kinasa

30
Déficit de G6PDH Es la enzimopatía mas frecuente y se hereda ligada al sexo. La hemólisis de esta es fundamentalmente intravascular.

31
Patogenia de la hemólisis
La G6PDH es fundamental para la síntesis de glutatión reducido. Protege de la hemólisis por parte de agentes antioxidantes. La Hb se transforma en meta-Hb y en sulfo-Hb. Precipita en el interior del eritrocito dando cuerpos de Heinz.

32
Manifestaciones clínicas
Clínica solo en situaciones de estrés oxidativo, ellas son: Ingesta de antipiréticos (AAS), de antimaláricos(primaquina), antibióticos (isoniazida), ingesta de habas que son ricas en L-dopa, y un metabolito de ella es un potente oxidante otras circunstancias son las infecciones, la cetoacidosis diabética.
, de antimaláricos(primaquina), antibióticos (isoniazida), ingesta de habas que son ricas en L-dopa, y un metabolito de ella es un potente oxidante. otras circunstancias son las infecciones, la cetoacidosis diabética.")
33
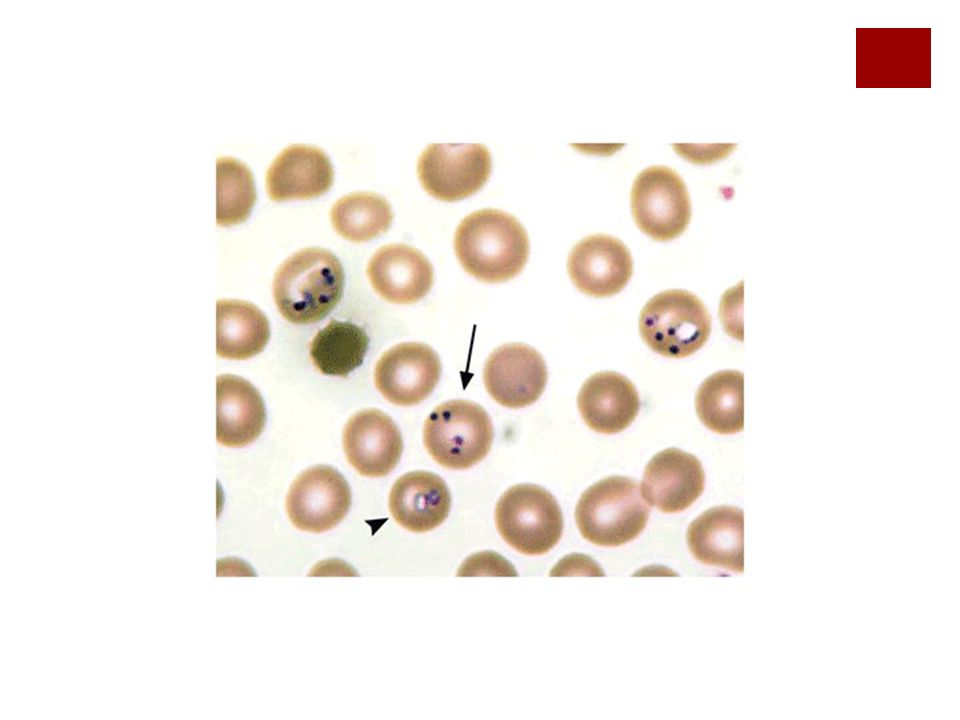
Clínica de déficit de G6PDH
Anemia Hemoglobinuria Haptoglobina baja Dolor lumbar Corpúsculos de Heinz Además, hay mayor propensión a padecer infecciones por que se produce una alteración en la bacteriolisis

34
Tratamiento Evitar situaciones de estrés oxidativo
Si aparecen crisis hemolíticas graves hay que transfundir hematíes Hidratar correctamente al paciente Alcalinizar la orina (para evitar precipitados)
")
35
Déficit de Piruvato cinasa
Alteración en la vía glicolitica con déficit de ATP. Aumenta los metabolitos intermedios se van para la vía del 2,3 difosfoglicerato con lo que aumenta la liberación de oxigeno a los tejidos por parte de la Hb y el grado de anemia es menor a lo esperado.

36
Déficit de piruvato cinasa
Hemólisis extravascular. Actividad del piruvato cinasa está disminuido en el hemolizado. Aumento del 2,3 difosfoglicerato. En el frotis no se observan esferocitos y la fragilidad osmótica es normal. Tx ac fólico

37
Anemias hemolíticas por alteración en la síntesis de Hb
Existen dos grupos de alteraciones: Las que afectan a la estructura de la Hb Los defectos en la síntesis de una cadena globinica que se denominan Talasemias.

38
Estructura y función de la Hb
Un hematíe contiene aproximadamente 600 mill de moléculas de Hb. Cada molécula de HbA esta formada por 4 cadenas polipeptidicas(2α y 2β) cada una a su vez contiene su propio hem los tipos de Hb en el adulto son: HbA(95-98%), HbA2 (1-3%), HbF(1%)
 cada una a su vez contiene su propio hem. los tipos de Hb en el adulto son: HbA(95-98%), HbA2 (1-3%), HbF(1%)")
39
Hemoglobinopatia y Talasemia
Hemoglobinopatía Alteración de un aminoácido Alteración de una cadena de globina

40
Hemoglobinopatías Anemia de células falciformes Rasgo falciforme
Estados heterocigóticos dobles Hemoglobinas inestables Hb de alta afinidad por el oxígeno (eritrocitosis familiar) Metahemoglobinemia (cianosis familiar)
 Metahemoglobinemia (cianosis familiar)")
41
Anemia de células falciformes
También se llama anemia drepanocítica o hemoglobinopatía S la alteración molecular consiste en la sustitución del ac glutámico en posición 6 de la cadena ß de la HbA normal por valina. Esta alteración hace que no se sintetice HbA sino HbS.

42
Anemia de células falciformes
Los eritrocitos con HbS son resistentes a la infección por Plasmodium vivax. Por ello, los sujetos con HbS en zonas endémicas como Africa, la mutación este frecuente en un 30-40% de la población.

43
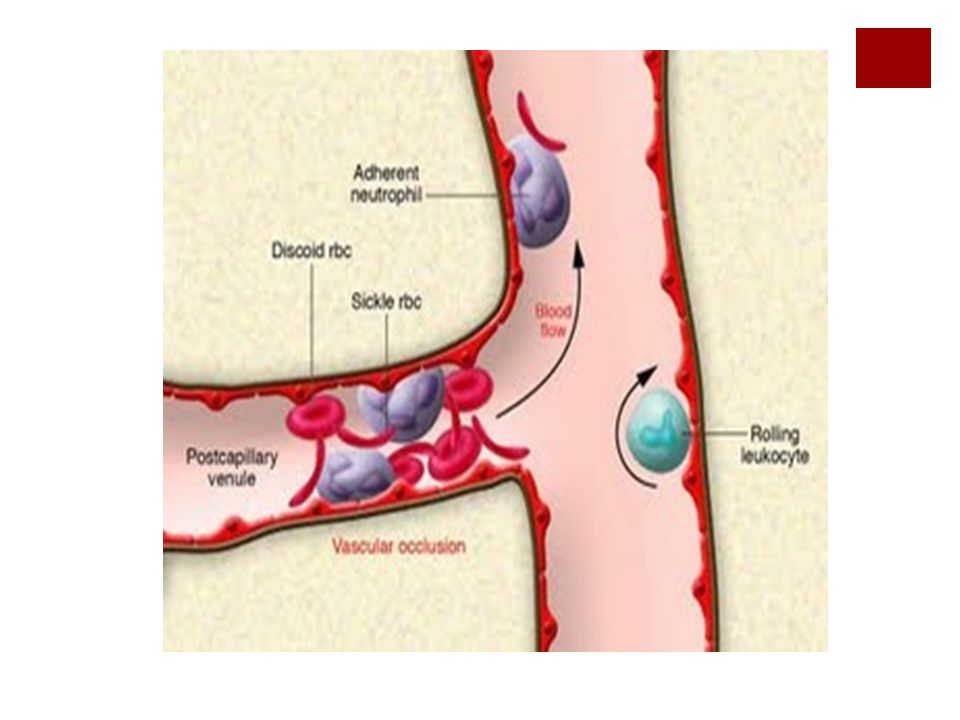
Patogenia y clínica A consecuencia de la mutación, cuando se desoxigena la Hb esta se polimeriza formando los llamados cuerpos tactoides que se distribuyen cambiando la conformación del hematíe (hematíe falciforme). La clínica deriva de la obstrucción de los vasos terminales lo cual provoca:
. La clínica deriva de la obstrucción de los vasos terminales lo cual provoca:")
45
Patogenia y clínica Un síndrome hemolítico, dolor e infartos,
La clínica es la de una anemia crónica con episodios intercalados de crisis hemolíticas y vaso oclusivas desencadenadas por infecciones, deshidratación, frío. Son frec las infecciones por encapsulados debido al hipoesplenismo y la osteomielitis por Salmonella.

46
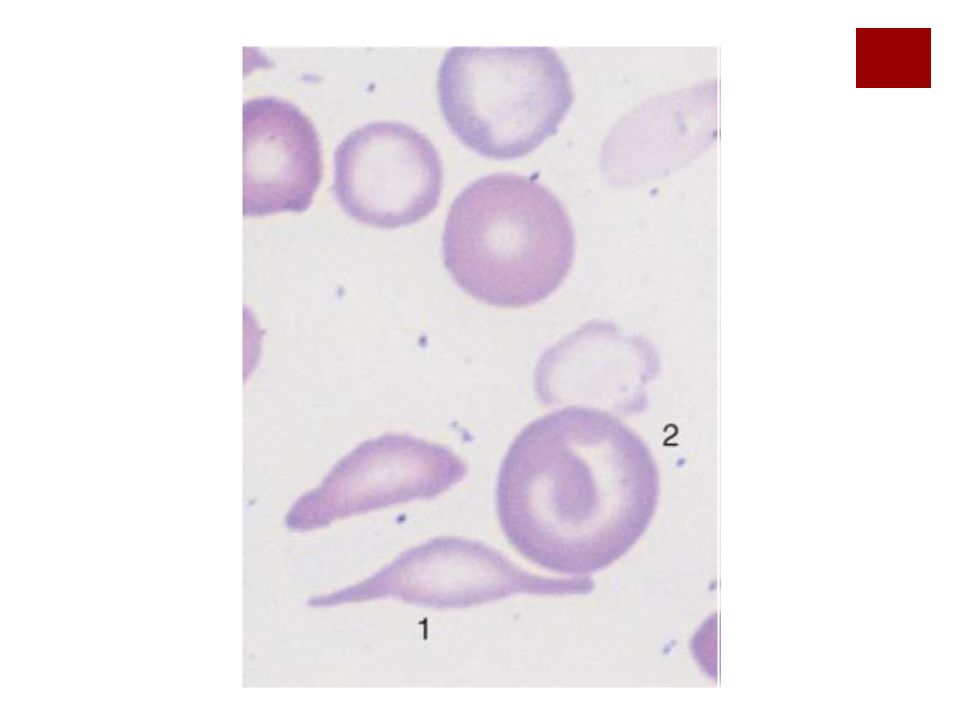
Diagnóstico La clínica
La electroforesis de Hb (la HbS tiene menor movilidad electroforética que la HbA por el test de falciformación Datos de sínd hemolítico En frotis de sangre se ven drepanocitos

52
Tratamiento Anemia: Transfusión y ac fólico, fármacos que aumenten la HbF (hidroxiurea), eritropoyetina, transplante alogénico de progenitores hematopoyéticos en casos graves Dolor: analgésicos e hidratación Prevención de infecciones con antibióticos de amplio espectro y vacuna anti neumocóccica Evitar climas muy fríos o calientes, otros
, eritropoyetina, transplante alogénico de progenitores hematopoyéticos en casos graves. Dolor: analgésicos e hidratación. Prevención de infecciones con antibióticos de amplio espectro y vacuna anti neumocóccica. Evitar climas muy fríos o calientes, otros.")
53
Talasemias Son aquellas alteraciones de la molécula de Hb que se produce por la falta de síntesis total o parcial de las cadenas de globinas. Cada tipo recibe el nombre de la cadena que deja de sintetizarse: p.e ß-talasemia.

54
Tipos de ß-talasemias ß talasemia menor ß talasemia intermedia
ß talasemia mayor

55
ß talasemia menor Es muy importante descubrirla y diferenciarla de la anemia ferropenica ya que es un error grave tratar a con hierro a un talasemico Hay ligera hepatoesplenomegalia, crisis de ictericia, ligero cansancio. La Hb esta en g/dl El VCM bajo(60-65 fL), la ADEsuele ser normal
, la ADEsuele ser normal.")
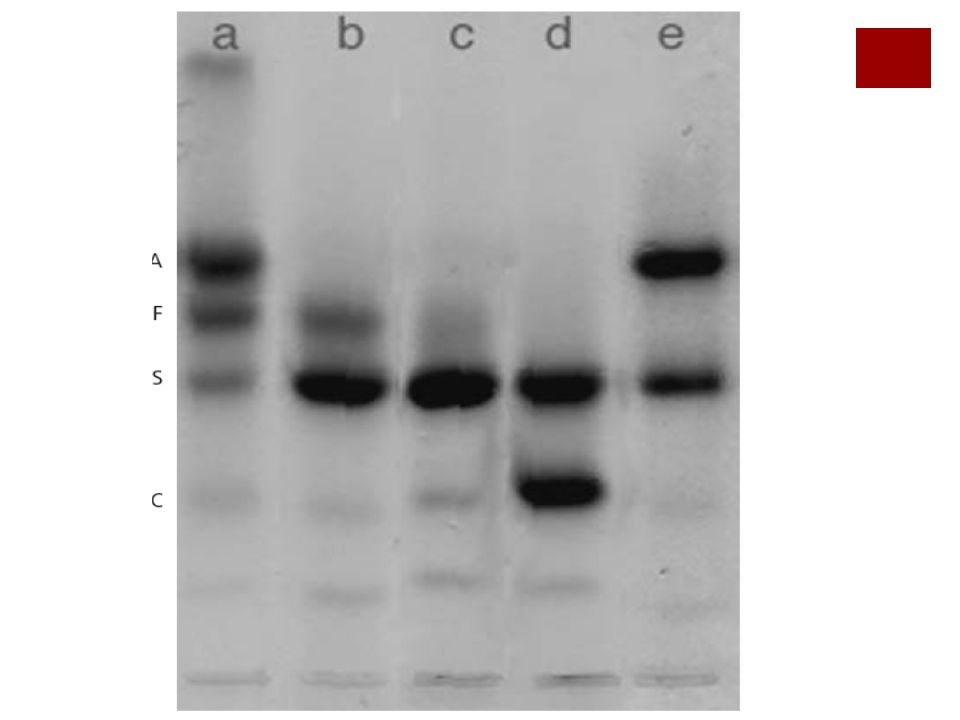
56
ß talasemia menor En la electroforesis de la Hb se detectan niveles elevados de HbA2 y en ocasiones HbF, dado que estos dos tipos no producen cadenas ß para su formación.

57
Anemias hemolíticas de origen inmune
Son de origen adquirido que se producen por anticuerpos frente a antígenos eritrocitarios. Estos anticuerpos los puede producir el mismo individuo frente a estructuras antigénicas propias (autoinmunes) o Sintetizarlos frente a antígenos de otro individuo (isoinmunes)
 o. Sintetizarlos frente a antígenos de otro individuo (isoinmunes)")
58
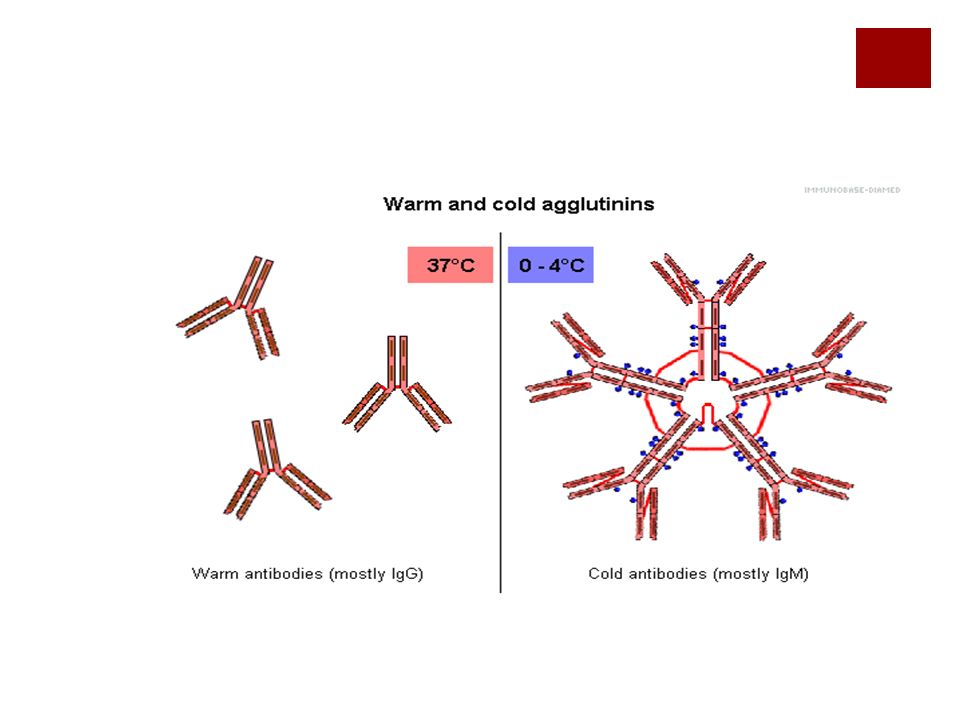
Anemias hemolíticas autoinmunes
Dos variedades: por anticuerpos calientes:la reacción se realiza a 37°C y el anticuerpo es IgG por anticuerpos fríos: la reacción es a baja temperatura y el anticuerpo suele ser IgM la causa de auto-anticuerpos a veces se desconoce.

59
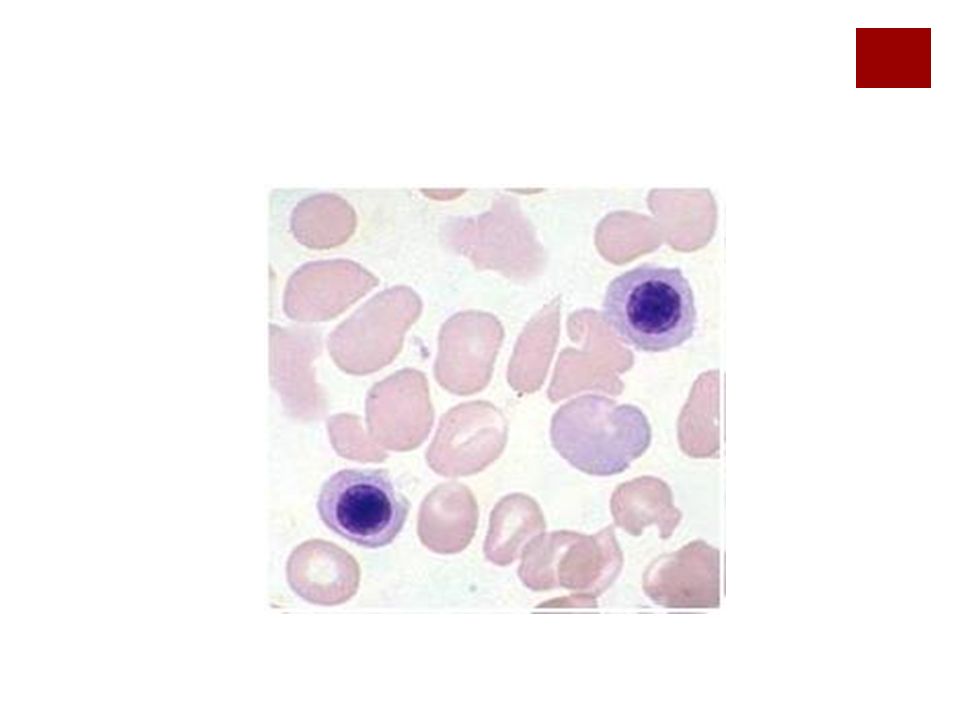
Test de Coombs El directo permite detectar anticuerpos en la superficie de las células y El indirecto detectar anticuerpos en el suero del paciente.

60
Test de Coombs directo Positivo en: AHAI EHRN RHPT

63
Test de Coombs directo Anemia Hemolítica Acs. calientes Anti IgG +
Anti C3 + Anemia Hemolítica Acs. Fríos Anti C3 +

64
Test de Coombs indirecto
Se incuba suero del enfermo con hematíes de fenotipo conocido. Si el enfermo posee alo o auto anticuerpos sensibilizará los hematíes control y al añadir suero de coombs se producirá aglutinación.

65
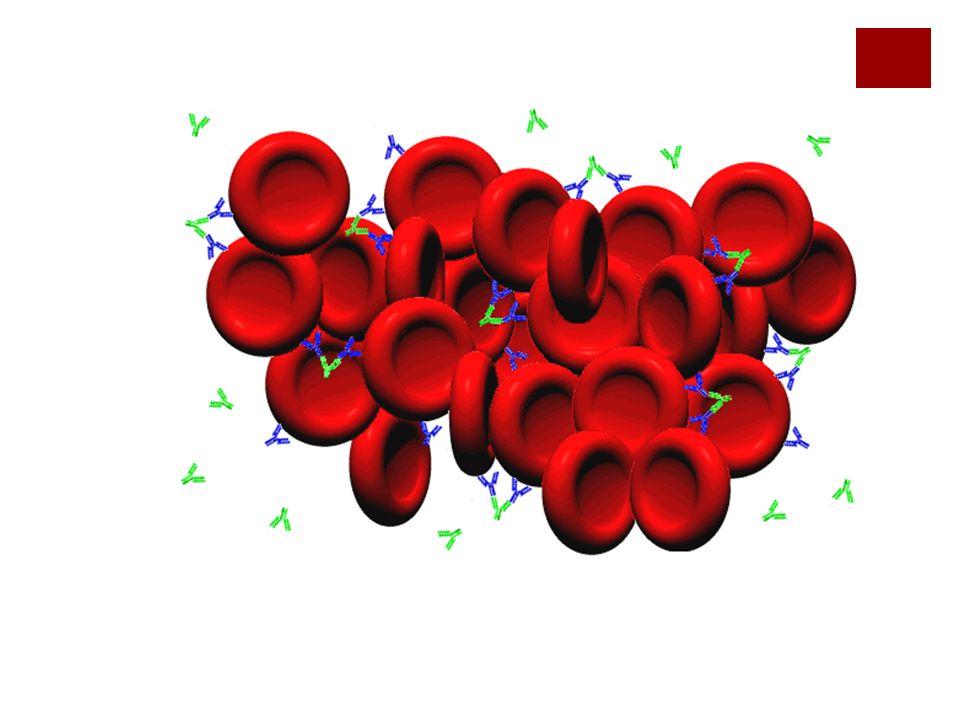
Patogenia Los auto anticuerpos IgG pueden
Activar el complemento hasta el final(C5-C9), tendremos hemólisis intravascular La activación se frene a nivel del C3 y en este caso los hematíes van a ser capturados por macrófagos hepáticos (extravascular) Formación de esferocitos.
, tendremos hemólisis intravascular. La activación se frene a nivel del C3 y en este caso los hematíes van a ser capturados por macrófagos hepáticos (extravascular) Formación de esferocitos.")
66
Causas de AH por anticuerpos calientes
Sind linfoproliferativos Enf autoinmunes y colagenopatías Tumores sólidos Infecciones víricas 20-50% idiopáticas a veces se asocian a púrpura trombocitopénica idiopática S Evans

67
Clínica y biología Existen dos formas:
Agudas: hay sínd hemolítico, aumento de bilirrubina indirecta, haptoglobina baja y hemoglobinuria, es test de coombs directo es positivo en 95% y 2/3 tienen el indirecto positivo. Crónicas: con hepatoesplenomegalia en el 50% de los casos.

68
Tratamiento Corticoides a dosis altas
Esplenectomía, pero no en niños menores de 6 años por el riesgo de infecciones por cocos G+ Ig iv Inmunosupresores

69
Anemia hemolítica por anticuerpos fríos
Producidas por: Mononucleosis infecciosa. Infección por Micoplasma pneumoniae. Sínd linfoproliferativos. Infección por Treponema Idiopática

70
Diagnóstico Por la clínica: acrocianosis en capilares distales, con dolor, y sind hemolítico. Por datos biológicos: anemia de intensidad variable, aumento de reticulocitos, test de coombs positivo para el complemento pero negativo para IgG, crioaglutininas aumentadas. La aglutinación desaparece al calentar la sangre a 37°C

71
Tratamiento Calentar las partes acras y evitar la exposición al frío
Transfusión de hematíes a 37°C Administrar Ig polivalentes a dosis altas Plasmaféresis

72
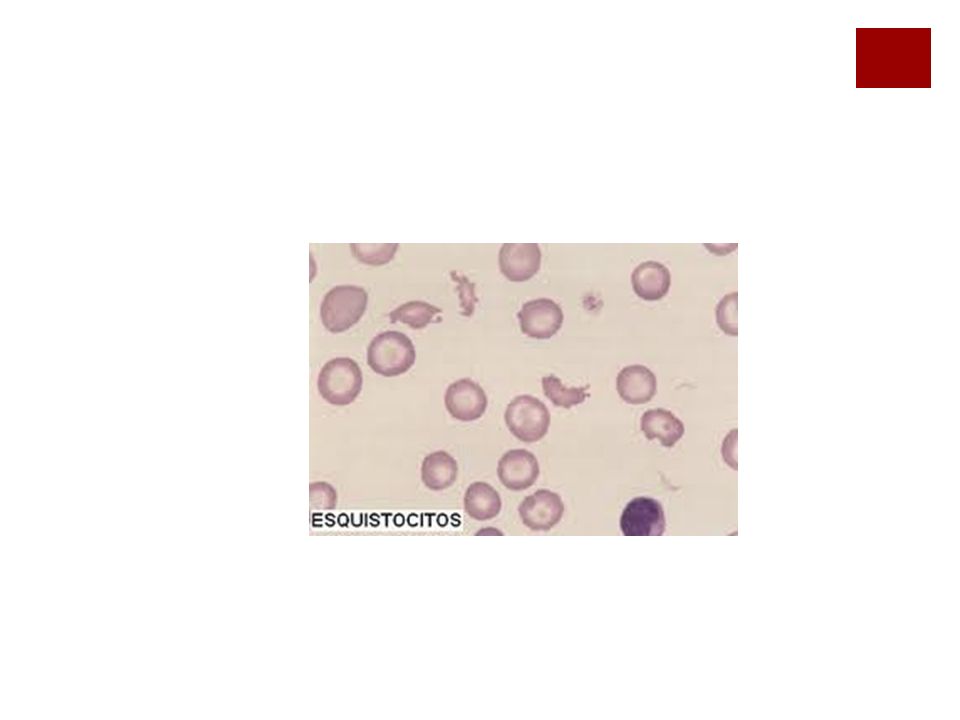
Algunas anemias de mecanismo complejo
Síndrome de Zieve Anemia hemolítica microangiopática Anemia hemolítica macroangiopática por acción directa de agentes infecciosos por tóxicos por venenos de animales

74
Hemoglobinuria paroxística nocturna
Es una anemia hemolítica asociado a un fallo medular, que se debe a un defecto genético por el que los hematíes se hacen mas susceptibles a la lisis por complemento. Además con riesgo aumentado de trombosis

75
Cuadro clínico Un cuadro de anemia hemolítica intravascular con hemoglobinuria. La hemólisis se produce mas por la noche por que el pH de la sangre es mas ácido lo que facilita la activación del complemento, la orina es oscura color coca cola, es frecuente la ferropenia, pueden tener trastornos trombóticos así como crisis de aplasia

76
Diagnóstico Por clínica Por estudios de biología molecular
Inmunofenotipo Test clásicos: prueba de Ham-Dacie o la prueba de la sucrosa.

77
Pronóstico y tratamiento
Pronóstico: es variable depende de las complicaciones. Tratamiento: ácido fólico Hierro transfusión frente al clon anormal: inmunosupresores, transplante alogénico de precursores hematopoyéticos (casos graves porque puede provocar la muerte en procedimiento)
")
78
GRACIAS

Presentaciones similares













>")




 ESPECÍFICAS (Respuesta inmunitaria) – La unión antígeno anticuerpo es específica.>")



