Descargar la presentación
La descarga está en progreso. Por favor, espere

1
Leucemia mieloide crónica y neoplasias mieloproliferativas crónicas ph neg
Dr. Juan Richmond N. Especialista en Medicina Interna/Hematología Servicio de Hematología Hospital Calderón Guardia

2
Introducción Definición:
Conjunto de entidades hematológicas malignas con características clínicas y evolutivas muy afines, y de etiopatogenia probablemente común. Caracterizados por la expansión clonal de una célula madre (stem cell) pluripotente, dando como resultado una hipercelularidad medular con predominio de una línea específica y con posibilidad de evolucionar a transformación leucémica. Son alteraciones neoplásicas que afectan a la serie mieloide.
 pluripotente, dando como resultado una hipercelularidad medular con predominio de una línea específica y con posibilidad de evolucionar a transformación leucémica. Son alteraciones neoplásicas que afectan a la serie mieloide.")
3
Introducción CONCEPTO: Procesos de evolución crónica con panmielosis.
Curso crónico inicialmente. Habitualmente esplenomegálicos. Cursan con recuentos hemáticos aumentados Poliglobulia, leucocitosis, trombocitosis Frecuente eosinofilia, basofilia.

4
Introducción CONCEPTO:
Pueden producir hematopoyesis extramedular (bazo, hígado) Cursan con: Hiperuricemia Aumento de LDH y vitamina B12 Alteraciones en la FAG (FAL) Esplenomegalia Tendencia a la hemorragia Cierto grado de fibrosis medular
 Cursan con: Hiperuricemia. Aumento de LDH y vitamina B12. Alteraciones en la FAG (FAL) Esplenomegalia. Tendencia a la hemorragia. Cierto grado de fibrosis medular.")
5
Introducción Neoplasias Mieloproliferativas crónicas:
Leucemia Mieloide Crónica (LMC) Ph positivo Neoplasias mieloproliferativos crónicas Ph negativo Policitemia Vera (PV) Trombocitemia Esencial (TE) Mielofibrosis Idiopática (MF)
 Ph positivo. Neoplasias mieloproliferativos crónicas Ph negativo. Policitemia Vera (PV) Trombocitemia Esencial (TE) Mielofibrosis Idiopática (MF)")
6
Clasificación de las hemopatías malignas OMS 1999
Introducción Clasificación de las hemopatías malignas OMS 1999 Enfermedades mieloproliferativas LMC con cromosoma Filadelfia positivo Leucemia neutrofílica crónica Leucemia eosinofílica crónica/ Síndrome hipereosinofílico Mielofibrosis idiopática crónica Policitemia Vera Trombocitemia esencial Enfermedad mieloproliferativa, no clasificable Enfermedades mielodisplásicas/mieloproliferativos Leucemia mielomonocítica crónica Leucemia mielomonocítica juvenil Leucemia mieloide crónica atípica

7
Hematology, Jan 2006; 2006:

8
LEUCEMIA MIELOIDE CRÓNICA

9
Leucemia Mieloide Crónica
Concepto: Es el SMPC de mayor importancia clínica Por su frecuencia y pronóstico Proliferación de carácter clonal originada en la célula madre (stem cell) pruripotente común a las tres series hematopoyéticas. Intensa proliferación de la serie granulocítica en la MO, SP y otros órganos (bazo). Leucocitosis intensa, representados todos los elementos madurativos de la granulopoyesis, acompañada de esplenomegalia y disminución de la FAL.
 pruripotente común a las tres series hematopoyéticas. Intensa proliferación de la serie granulocítica en la MO, SP y otros órganos (bazo). Leucocitosis intensa, representados todos los elementos madurativos de la granulopoyesis, acompañada de esplenomegalia y disminución de la FAL.")
10
Leucemia Mieloide Crónica
Concepto: La mayoría de los pacientes presentan anomalía cromosómica en la MO denominada Cromosoma Filadelfia o Ph, t(9;22) Gen bcr/abl debido al intercambio genético de los cromosomas 9 y 22 en las células hematopoyéticas. No es exclusiva de la LMC 20% LLA de la infancia y < 3% de las LMA.
 Gen bcr/abl debido al intercambio genético de los cromosomas 9 y 22 en las células hematopoyéticas. No es exclusiva de la LMC. 20% LLA de la infancia y < 3% de las LMA.")
11
Leucemia Mieloide Crónica

12
Leucemia Mieloide Crónica

13
Leucemia Mieloide Crónica

14
Leucemia Mieloide Crónica
Etiología y epidemiología: Etiología desconocida. Aumento en la incidencia en radiaciones o benceno Curso evolutivo bifásico Fase inicial o crónica Fase final o crisis blástica, similar a una leucemia aguda Muchos pacientes se intercalan con un tercer periodo, la fase de aceleración.

15
Leucemia Mieloide Crónica
Etiología y epidemiología: Representa el 15 – 20% de todas las leucemias. Incidencia 1,5 casos/ hab/año. Puede aparecer a cualquier edad pero es raro en la infancia Predomina en las edades media y avanzada de la vida Mediada de edad al momento del dx: 50 años Predomina ligeramente en varones. Incidencia familiar es excepcional.

16
Leucemia Mieloide Crónica
Manifestaciones Clínicas: Asintomáticos (fortuito). Se diagnóstica en el 95% de los casos en la fase crónica. Cuyo inicio es habitualmente insidioso. El diagnóstico es precedido Sxs inespecíficos (astenia, adinamia, pérdida de peso, sudoración nocturna, fibrícula) Relacionado con un estado de hipermetabolismo Molestias provocadas por la esplenomegalia o infarto esplénico. Otros síntomas: dolores óseos, litiasis renal, crisis de gota o priapismo, son menos frecuentes.
. Se diagnóstica en el 95% de los casos en la fase crónica. Cuyo inicio es habitualmente insidioso. El diagnóstico es precedido. Sxs inespecíficos (astenia, adinamia, pérdida de peso, sudoración nocturna, fibrícula) Relacionado con un estado de hipermetabolismo. Molestias provocadas por la esplenomegalia o infarto esplénico. Otros síntomas: dolores óseos, litiasis renal, crisis de gota o priapismo, son menos frecuentes.")
17
Leucemia Mieloide Crónica
Manifestaciones Clínicas: 5% se presente en crisis blástica inicial. La fase crónica pasa inadvertida. Datos indicativos de LMC Esplenomegalia marcada Basofilia Mielemia Trombocitosis El diagnóstico se confirmara por el estudio citogenético de la MO o molecular por TR-PCR.

18
Leucemia Mieloide Crónica
Clínica: Al examen físico El hallazgo más frecuente Esplenomegalia (80-90%) Existe correlación entre la intensidad de la leucocitosis y el tamaño del bazo.
 Existe correlación entre la intensidad de la leucocitosis y el tamaño del bazo.")
19
Leucemia Mieloide Crónica
Laboratorio: Sangre periférica Leucocitosis de intensidad variable ( – /ml) 30% cursan con leucocitosis moderada /ml Leucocitosis granulocítica, representando todos los estadios madurativos de la granulopoyesis, con predominio de las formas maduras. Mayor proporción mielocitos que de metamielocitos Porcentaje de blastos en SP es escaso o nulo La presencia de blastos debe hacer sospechar fase blastica o acelerada La basofilia es constante, es más rara la eosinofilia.
 30% cursan con leucocitosis moderada /ml. Leucocitosis granulocítica, representando todos los estadios madurativos de la granulopoyesis, con predominio de las formas maduras. Mayor proporción mielocitos que de metamielocitos. Porcentaje de blastos en SP es escaso o nulo. La presencia de blastos debe hacer sospechar fase blastica o acelerada. La basofilia es constante, es más rara la eosinofilia.")
20
Leucemia Mieloide Crónica

21
Leucemia Mieloide Crónica
Laboratorio: Sangre periférica En un 50% existen eritroblastos circulantes Es frecuente la anemia moderada. Cifra de plaquetas normal o aumentada (trombocitosis 45%) Tendencia al sangrado por alteración en el funcionalismo plaquetario. Disminución de la actividad de la FAL, a menudo es de 0. Uno de los datos más típicos de la LMC AMO: intenso incremento en la celularidad hemopoyética, sobre todo de la serie granulocítica.
 Tendencia al sangrado por alteración en el funcionalismo plaquetario. Disminución de la actividad de la FAL, a menudo es de 0. Uno de los datos más típicos de la LMC. AMO: intenso incremento en la celularidad hemopoyética, sobre todo de la serie granulocítica.")
22
Leucemia Mieloide Crónica
Laboratorio: Estudio citogenético Revela la presencia del cromosoma Ph en el 95% de los casos. Otros datos de laboratorio Aumento constante de la Vit B12 y de sus proteínas transportadoras (transcobalamina) Aumento de la LDH Aumento menos frecuente del ácido úrico.
 Aumento de la LDH. Aumento menos frecuente del ácido úrico.")
23
Leucemia Mieloide Crónica
Evolución y pronóstico La mayoría se diagnóstica en la fase crónica Al cabo de una mediana de 4 años la enfermedad entra en una fase más agresiva Resistente habitualmente al tratamiento Crisis blástica de la LMC En muchos pasa de forma brusca, en otros pasa antes por una denominada fase de aceleración.

24
Leucemia Mieloide Crónica
Evolución y pronóstico Fase de aceleración Se da en un 40% de los casos Cambios en las características clínicas y hematológicas de la enf. Fiebre, sudoración, pérdida de peso, dolores óseos, crecimiento progresivo del bazo a pesar del ajuste en el tratamiento Suele acompañarse de alteraciones hematológicas Anemia, trombocitopenia o trombocitosis intensa, basofilia marcada (>20%), blastosis en sangre periférica o médula ósea mayor de los habitual No alcanza valores de Crisis Blástica
, blastosis en sangre periférica o médula ósea mayor de los habitual. No alcanza valores de Crisis Blástica.")
25
Leucemia Mieloide Crónica
Evolución y pronóstico Crisis Blástica Es una autentica leucemia aguda Los que no pasan por una fase de aceleración se asiste la invasión rápida de la SP, la MO y a veces otros órganos por células blásticas. Rápido deterioro del paciente Aparece blástosis periférica y medular

26
Leucemia Mieloide Crónica
Evolución y pronóstico Sin tratamiento sobrevida media de 2,5 años Algunos fallecen en la fase crónica por causas no relacionadas con la enfermedad. Después del dx de crisis blástica Sobrevida de 4 a 5 meses.

27
POLICITEMIA VERA

28
Policitemia Vera Etiopatogenia Enfermedad neoplásica.
Resultado de la proliferación anormal de una célula madre pluripotente, que da lugar a una hemopoyesis clonal de hematíes, granulocitos y plaquetas, predominando, con mucho, la hiperplasia eritroide. No afecta progenitores de células T y NK

29
Policitemia Vera Etiopatogenia
Hipersensibilidad de los precursores eritroides a la acción de la EPO. También una capacidad de formación de colonias Epo-independientes.

30
Policitemia Vera Etiopatogenia
También se ha observado, valores elevados de Bcl-xL Proteína inhibitoria de la apoptosis Explica que estas colonias pueden sobrevivir sin EPO El receptor de la TPO (Mpl) se haya disminuido Sugiere que la proliferación y diferenciación de los megacariocitos en la PV es independiente de TPO. Hipersensibilidad a los factores de crecimiento y citocinas (IL-3, Epo, GM/CSF). Es una enfermedad acumulativa no invasiva.
 se haya disminuido. Sugiere que la proliferación y diferenciación de los megacariocitos en la PV es independiente de TPO. Hipersensibilidad a los factores de crecimiento y citocinas (IL-3, Epo, GM/CSF). Es una enfermedad acumulativa no invasiva.")
31
Policitemia Vera Manifestaciones clínicas
Incidencia 0,8-1,5 casos/ hab/año Mediana de edad al momento del Dx ♂ 60 años y ♀ 62 años Relación ♂:♀ (1,2: 1) 5-7% tienen menos de 40 años
 5-7% tienen menos de 40 años.")
32
Policitemia Vera Manifestaciones clínicas
15-25% se diagnostica en forma casual. 0,4% tienen historia familiar de PV La mayoría de las manifestaciones son secundarias a la proliferación excesiva de las diferentes líneas celulares.

33
Signos y Síntomas en la Policitemia Vera
% Signos Cefaleas Astenia Molestias epigástricas Mareo Alteraciones Visuales Disnea Prurito Sudoración Adelgazamiento Parestesias Angor Gota Epistaxis 41-48 35-37 23-50 25-43 10-31 23-34 14-43 33 16-29 29 15-23 4-26 17 Plétora Inyección conjuntival Esplenomegalia Hepatomegalia Hipertensión Úlceras cutáneas 65-84 28-59 50-81 31-50 2

34
Policitemia Vera Complicaciones trombóticas
Principal causa de morbilidad y mortalidad 14% presentan eventos trombóticos antes del Dx La mayoría 2 años antes, gnlm IAM, ECV isquémico 20% como 1ra manifestación de la enfermedad. 66% son trombosis arteriales 70% ICTUS e ICT

35
Policitemia Vera Complicaciones trombóticas durante la enfermedad
Trombosis arterial más frecuente (50%) Venosas 38% Las complicaciones más frecuentes IAM y ECV 81% trombosis mortales Incidencia de trombosis 3,4%/año Mortalidad global 2,9/100 ptes/año
 Venosas 38% Las complicaciones más frecuentes IAM y ECV. 81% trombosis mortales. Incidencia de trombosis 3,4%/año. Mortalidad global 2,9/100 ptes/año.")
36
Policitemia Vera Trombosis venosas
Más frecuentes en venas profundas de MsIs Complicándose con TEP También: Vena esplénica, hepática o mesentérica Síndrome de Budd Chiari PV causa del 10% de Síndrome de Budd Chiari Ocasiona HT portal, dolor abdominal, hepatosplenomegalia, ascitis, edemas en MsIs, ictericia y distención de venas abdominales.

37
Policitemia Vera Otras manifestaciones vasculares
Eritromelalgía Isquemia digital con pulsos palpables Tromboflebitis Factores de riesgo para complicaciones vasculares Hto principal determinante Incremento de la viscosidad sanguínea Disminución del flujo periférico Debe mantenerse Hto < 46%

38
Policitemia Vera

39
Policitemia Vera Complicaciones hemorrágicas
30-40% presentan algún tipo de manifestación hemorrágica Hemorragias GI (más frecuente úlceras gástricas) Hemorragias de Várices esofágicas Hemorragias cerebrales La predisposición hemorrágica Se atribuye a alteraciones cualitativas de las Pks
 Hemorragias de Várices esofágicas. Hemorragias cerebrales. La predisposición hemorrágica. Se atribuye a alteraciones cualitativas de las Pks.")
40
Policitemia Vera Laboratorio Dato más destacado
Gran aumento de la cifra de hematíes Suele superar los 6 x 1011/dl La cifra de Hb también se halla elevado superando los 20g/dl Hto >60% se detecta en algo más del 50%

41
Policitemia Vera Laboratorio Anomalías eritrocitarias
Microcitosis e hipocromia por déficit de hierro. También presentan leucocitosis y trombocitosis 2/3 de los ptes tienen más de un 3% de basofilos Presencia ocasional de formas granulocitarias inmaduras

42
Policitemia Vera Laboratorio AMO:
La cifra de pks superior a /dl 40-50% Superior a 1 millón/dl en el 10% de los casos AMO: Hipercelular, hiperplasia de la serie eritroide, granulocítica y megacariocítica Hiposiderosis: ferropenia por hiperconsumo (95%) Importante incremento de la FAL Índice suele hallarse por encima de 100
 Importante incremento de la FAL. Índice suele hallarse por encima de 100.")
43
Policitemia Vera Bx Medular:
Hipercelular. Concentración sérica de Vit B12 elevada en el 40% de los casos. Transcobalamina 70% LDH puede estar elevada Se incrementa más cuando evoluciona a metaplasia mieloide pospolicitémica. Ferritina sérica disminuida.

44
Policitemia Vera Cultivo in vitro de progenitores hematopoyéticos
Crecimiento de colonias eritroides en ausencia de Epo, fenómeno denominado crecimiento endógeno o espontáneo.

45
Policitemia Vera Diagnóstico:
POLICITEMIA ABSOLUTA o masa eritrocitaria aumentada: Es la citemia que excede más del 25% el valor normal calculado para el paciente. La única manera del hacer el diagnóstico, es con un estudio isotópico de volúmenes: citemia, plasmemia y volemia. Siempre tienen una Poliglobulia Absoluta Varón con Hto > 60% Mujer con Hto > 55%

46
Policitemia Vera Diagnóstico: POLICITEMIA RELATIVA:
Se define como un incremento en el Hto causado por una disminución del volumen plasmático, con una citemia con valores normales. Policitemia Vera inaparente: Incremento del volumen plasmático que enmascara el de la citemia, pareciendo aparentemente normales a las cifras de Hb y Hto. Se observa en dos situaciones: Incremento del volumen plasmático como HTportal Cuando existe ferropenia, que determina una disminución sustancial de la concentración de Hb.

47
Policitemia Vera Diagnóstico:
The Polycythemia Vera Study Group (PVSG) estableció en 1975 criterios diagnósticos para PV. Estos criterios, aunque validos, pueden excluir ptes con manifestaciones moderadas o en fase inicial de la enfermedad. Estos criterios han sido desplazados por otros como: Cultivos in vitro de progenitores eritroides Marcadores de clonicidad La eritropoyetina Detección de esplenomegalia no palpable.
 estableció en 1975 criterios diagnósticos para PV. Estos criterios, aunque validos, pueden excluir ptes con manifestaciones moderadas o en fase inicial de la enfermedad. Estos criterios han sido desplazados por otros como: Cultivos in vitro de progenitores eritroides. Marcadores de clonicidad. La eritropoyetina. Detección de esplenomegalia no palpable.")
48
Criterios diagnósticos de la PV según PVSG
Policitemia Vera Criterios diagnósticos de la PV según PVSG Criterios Mayores Criterios Menores A1 Masa eritrocitaria Varones ≥ 36 ml/kg Mujeres ≥ 32 ml/kg A2 Sat O2 ≥ 92% A3 Esplenomegalia B1 Trombocitosis ≥ /dl B2 Leucocitosis ≥ 12 x 109/L B3 Índice FAL > 100 B4 Vit B12 sérica > 900 pg/ml o CTB12 > 2200 pg/ml Diagnóstico de PV si: A1+A2+A3 A1+A2+ 2 criterios de la categoría B

49
Criterios diagnósticos modificados de la PV
Policitemia Vera Criterios diagnósticos modificados de la PV A1 Aumento de la citemia (mayor del 25% del valor normal calculado) A2 Ausencia de causa de poliglobulia secundaria A3 Esplenomegalia palpable A4 Marcador de clonicidad (Ejem: cariotipo medular anómalo) B1 Trombocitosis > B2 Leucocitosis neutrofílica (neutrófilos > 10x109/L) B3 Esplenomegalia demostrada por US o Gammagrafía B4 Crecimiento de BFU-E característico o disminución de la EPO sérica Diagnóstico de PV: A1+A2+A3 o A4 A1+A2+ 2 criterios de categorïa B
 A2 Ausencia de causa de poliglobulia secundaria. A3 Esplenomegalia palpable. A4 Marcador de clonicidad (Ejem: cariotipo medular anómalo) B1 Trombocitosis > B2 Leucocitosis neutrofílica (neutrófilos > 10x109/L) B3 Esplenomegalia demostrada por US o Gammagrafía. B4 Crecimiento de BFU-E característico o disminución de la EPO sérica. Diagnóstico de PV: A1+A2+A3 o A4. A1+A2+ 2 criterios de categorïa B.")
50
Parámetros clínicos y de laboratorio asociados a PV
Policitemia Vera Parámetros clínicos y de laboratorio asociados a PV Poliglobulia importante. Leucocitosis persistente (con basofilia absoluta) Trombocitosis persistente* Microcitosis e Hipoferritinemia Esplenomegalia Prurito acuógeno Trombosis o historia previa de trombosis
 Trombocitosis persistente* Microcitosis e Hipoferritinemia. Esplenomegalia. Prurito acuógeno. Trombosis o historia previa de trombosis.")
51
Clasificación de las eritrocitosis
Primaria Congénita -Mutaciones del gen del receptor de la eritroyetina Adquirida -Policitemia Vera Secundaria -Hemoglobinopatía con alta afinidad por el O2. -Disminución del 2,3 DPG-eritrocitario -Hiperproducción de eritropoyetina -Hipoxia arterial Hipoxia de altitud Cardiopatías congénitas cianóticas Enfermedad pulmonar crónica Hipoventilación alveolar (Síndrome de Pickwick) -Disminución de la liberación de O2 tisular (tabaquismo) -Lesiones renales Hipernefroma Poliquistosis renal Quistes simples Nefropatías difusas Hidronefrosis Estenosis de la arteria renal Transplante renal -Lesiones hepáticas Hepatoma Cirrosis -Lesiones endocrinas Adenomas, tumores suprarrenales -Tumores varios Hemangioblastoma cerebeloso, carcinama bronquial y ovárico, fibromiomas -Fármacos Andrógenos Relativa (síndrome de Geisböck, Eritrocitosis espúrea o de estrés)
 -Disminución de la liberación de O2 tisular (tabaquismo) -Lesiones renales. Hipernefroma. Poliquistosis renal. Quistes simples. Nefropatías difusas. Hidronefrosis. Estenosis de la arteria renal. Transplante renal. -Lesiones hepáticas. Hepatoma. Cirrosis. -Lesiones endocrinas. Adenomas, tumores suprarrenales. -Tumores varios. Hemangioblastoma cerebeloso, carcinama bronquial y ovárico, fibromiomas. -Fármacos. Andrógenos. Relativa (síndrome de Geisböck, Eritrocitosis espúrea o de estrés)")
52
Diagnóstico diferencial de las poliglobulias
Policitemia Vera Poliglobulia secundaria Poliglobulia relativa Hematocrito Volumen eritrocitario Volumen plasmático Sat O2 Esplenomegalia Trombocitosis Leucocitosis Basofilia FAG Vitamina B12 Eritropoyetina Médula ósea BFU-E endógeno A N/A N Si D/N Hiperplasia global No No** Hiperplasia eritroide D A: aumentado, N: normal, D: disminuido, **posible en caso de tabaquismo

53
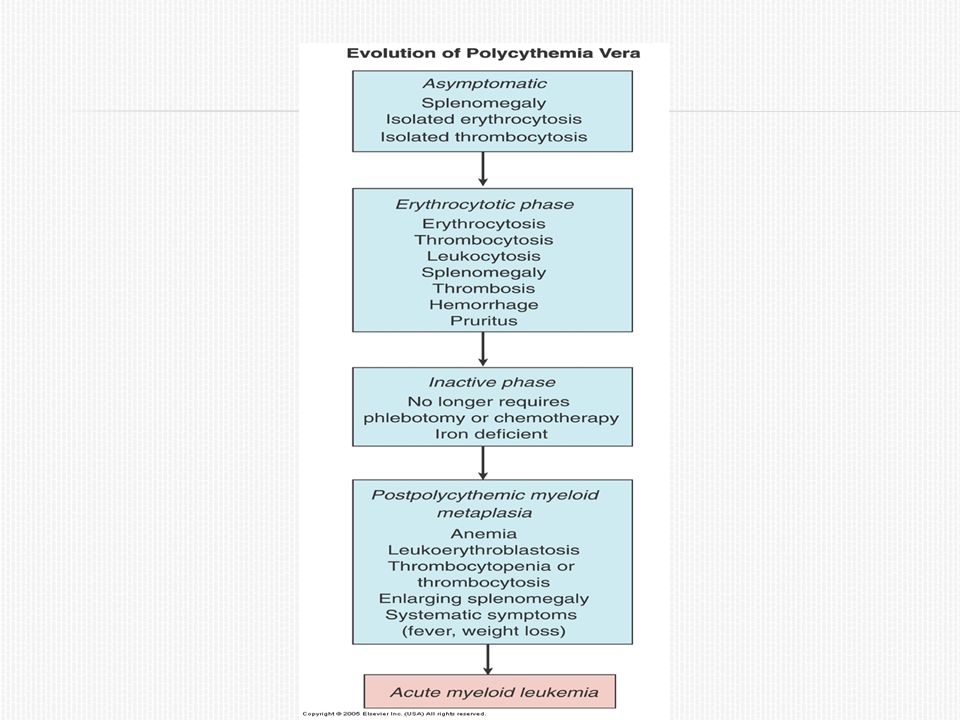
Policitemia Vera Evolución: Progresa por una serie de fases o estadios
Fase asintomática Fase sintomática Fase inactiva Fase de agotamiento: metaplasia mieloide pospolicitémica(mielofibrosis) 10% a los 10 a 15 años, 50% a los 20 años Incremento progresivo de la esplenomegalia, cuadro leucoeritroblástico con hematíes en lágrima, fibrosis medular difusa, normalización o disminución de la masa eritrocitaria, con anemia progresiva. LMA (20-50%)
 10% a los 10 a 15 años, 50% a los 20 años. Incremento progresivo de la esplenomegalia, cuadro leucoeritroblástico con hematíes en lágrima, fibrosis medular difusa, normalización o disminución de la masa eritrocitaria, con anemia progresiva. LMA (20-50%)")
55
TROMBOCITEMIA ESENCIAL
55

56
Trombocitemia esencial
Etiopatogenia SMPC caracterizado por un incremento persistente de la cifra de plaquetas y por una hiperplasia megacariocítica en la MO. Clínicamente se manifiesta por la tendencia a presentar complicaciones trombóticas y/o hemorrágicas Etiología es desconocida 1981, Fialkow demostró su clonicidad hematopoyética. 56

57
Trombocitemia esencial
Etiopatogenia Considerada hasta hace poco como la menos frecuente Actualmente es el SMPC más frecuente Incidencia 2-3 casos/ hab/año Mediana de edad al momento del diagnóstico 60 años Pico de incidencia 3ra y 4ta década de la vida 15-20% tienen menos de 40 años Enfermedad de predominio femenino (relación 1,6:1) Un tercio o más del total de enfermos se diagnostican de forma fortuita. 57
 Un tercio o más del total de enfermos se diagnostican de forma fortuita. 57.")
58
Trombocitemia esencial
Manifestaciones clínicas Exploración física Relativamente anodina, excepto 15-25% esplenomegalia. Principales causas de morbi-mortalidad Episodios trombóticos y/o hemorrágicos durante el curso de la enfermedad. Complicaciones hemostásicas Trombóticas (actualmente las más predominantes) Hemorrágicas Síntomas funcionales por alteración de la microcirculación. 58
 Hemorrágicas. Síntomas funcionales por alteración de la microcirculación. 58.")
59
Trombocitemia esencial
Manifestaciones clínicas Incidencias de complicaciones trombóticas del 31-83% La trombosis arterial es más frecuente que la venosa Cerebrovascular, vascular periférico y coronaria, y raramente grandes vasos arteriales y venosos. Complicaciones hemorrágicas Menos frecuentes que las trombóticas Afecta principalmente el aparato digestivo La púrpura equimótica y los hematomas son las manifestaciones cutáneas más frecuentes. Riesgo incrementado en ptes con Pks > 1,5 mill Se detecta biológicamente un déficit adquirido del factor de VW 59

60
Trombocitemia esencial
Manifestaciones clínicas Eritromelalgia Sensación de quemazón, dolor intenso,y enrojecimiento de dedosde la mano o de la planta del pie. El calor intensifica los síntomas y el frío los alivia. Sin tratamiento puede progresar a acrocianosis e incluso gangrena Se trata de un fenómeno trombótico intravascular causado por la activación y agregación plaquetaria en las arteriolas distales de las extremidades. La AAS revierte completamente el fenómeno inflamatorio y el dolor, restaurando la circulación periférica. 60

61
Trombocitemia esencial
Datos de laboratorio Hallazgos hematológicos al inicio de la enfermedad Normalidad de la Hb y del Hto Leucocitosis moderada ocasionales formas mieloides inmaduras Basofilia no mayor del 3% Agregados plaquetarios, anisocitosis de Pks, pks gigantes, vacuolización e hipogranularidad. 61

62
Trombocitemia esencial
Datos de laboratorio AMO: evidencia intensa hiperplasia megacariocitica Biopsia de Médula ósea Celularidad moderadamente aumentada con conservación del tejido adiposo Hiperplasia megacariocitica Trama reticulina normal o discretamente aumentada Cariotipo acostumbra ser invariablemente normal Alteraciones citogenéticas raras (<5%) 62
 62.")
63
Trombocitemia esencial
Diagnóstico No dispone de un marcador biológico característico Su diagnóstico se basa en criterios de exclusión 63

64
Trombocitemia esencial
Criterios diagnósticos de TE según PVSG (1997) I. Recuento de plaquetas > /ml II. Hematocrito < 40% o masa eritrocitaria normal (V< 36ml/kg, M < 32 ml/kg) III. Hierro medular presente o ferritina sérica normal o VCM normal IV. Ausencia de cromosoma Ph o de reordenamientos del gen bcr/abl V. Fibrosis colágena de la médula ausente o < 1/3 del área de la biopsia , sin esplenomegalia ni síndrome leucoeritroblástico acompañante VI. Ausencia de evidencia morfológia o citogénetica de SMD VII. Ausencia de causa conocida de trombocitosis reactiva 64
 I. Recuento de plaquetas > /ml. II. Hematocrito < 40% o masa eritrocitaria normal (V< 36ml/kg, M < 32 ml/kg) III. Hierro medular presente o ferritina sérica normal o VCM normal. IV. Ausencia de cromosoma Ph o de reordenamientos del gen bcr/abl. V. Fibrosis colágena de la médula ausente o < 1/3 del área de la biopsia , sin esplenomegalia ni síndrome leucoeritroblástico acompañante. VI. Ausencia de evidencia morfológia o citogénetica de SMD. VII. Ausencia de causa conocida de trombocitosis reactiva. 64.")
65
Trombocitemia esencial
Diagnóstico diferencial de la TE Síndromes mieloproliferativos crónicos Policitemia Vera Leucemia mieloide crónica Mielofibrosis idiopática Síndromes mielodisplásicos Anemia refractaria sideroblástica Síndrome 5q- Trombocitosis reactivas Esplenectomia Anemia Ferropénica Anemias hemolíticas Infecciones Enfermedades inflamatorias crónicas Neoplasias epiteliales LH y LNH Cirugía Vincristina Rebote postratamiento en déficit de B12 o fólico 65

66
Trombocitemia esencial
Evolución La evolución a leucemia aguda en los pacientes que no reciben tx quimioterapeútico es muy infrecuente (<1%). Más frecuente a subtipos FAB M4 y M7 66
. Más frecuente a subtipos FAB M4 y M")
67
MIELOFIBROSIS IDIOPÁTICA
67

68
Mielofibrosis Fibrosis de la médula ósea:
Fibrosis secundaria: afección por diferentes procesos, entre los cuales destacan infecciones como las tuberculosis, tumores sólidos y diversas neoplasias hematológicas (EH, LNH, tricoleucemia, SMD, leucemias agudas, etc). Dicha mielofibrosis revierte al remitir el proceso que la origina. Rara vez es intensa para provocar hemopoyesis extramedular. 68
. Dicha mielofibrosis revierte al remitir el proceso que la origina. Rara vez es intensa para provocar hemopoyesis extramedular. 68.")
69
69

70
Mielofibrosis Hemopoyesis extramedular se puede observar ocasionalmente en individuos Han recibido radiaciones ionizantes Tuberculosis Carcinomatosis diseminada Osteopetrosis o enfermedad de Paget 70

71
Mielofibrosis MIELOFIBROSIS Ó METAPLASIA MIELOIDE AGNOGÉNICA
Fibrosis de la médula ósea primaria, de causa desconocida, acompañada de metaplasia mieloide de otros órganos (bazo, hígado). 71
. 71.")
72
Mielofibrosis Etiopatogenia Etiología desconocida
Enfermedad clonal con origen en la célula madre pluripotente común a las tres series. Los fibroblastos de la MO no forman parte de la proliferación neoplásica. La proliferación fibroblástica se debe fundamentalmente a la liberación intramedular de platelet-associated growth factor, Producida por los megacariocitos y almacenada en los gránulos alfa de las plaquetas. 72

73
Mielofibrosis Etiopatogenia Etiología desconocida
Transforming growth factor β (TGF-β) regula la síntesis de la matrix extracelular Calmodulina sustancia que actúa como mitógeno para los fibroblastos y cuyas concentraciones urinarias se hallan elevadas en los individuos con MMA. 73
 regula la síntesis de la matrix extracelular. Calmodulina sustancia que actúa como mitógeno para los fibroblastos y cuyas concentraciones urinarias se hallan elevadas en los individuos con MMA. 73.")
74
Mielofibrosis Cuadro clínico
Constituye menos del 15% del total de casos de SMPC Es la menos frecuente de las cuatro entidades En personas de edad avanzada Mediana de edad al diagnóstico 65 años Casi una cuarta parte de los ptes con MMA tienen menos de 55 años Sintomatología de inicio insidioso 74

75
Mielofibrosis Cuadro clínico Se reconocen tres orígenes fundamentales
La anemia (astenia, palidez, palpitaciones, disnea de esfuerzo, edemas, acúfenos). Estado de hipermetabolismo (febrícula, sudoración, pérdida de peso, anorexia, en ocasiones gota, o litiasis renal por hiperuricemia) Esplenomegalia (molestias en hipocondrio Izquierdo, dolor agudo por infartos esplénicos 75
. Estado de hipermetabolismo (febrícula, sudoración, pérdida de peso, anorexia, en ocasiones gota, o litiasis renal por hiperuricemia) Esplenomegalia (molestias en hipocondrio Izquierdo, dolor agudo por infartos esplénicos. 75.")
76
Mielofibrosis Cuadro clínico Existe tendencia al sangrado Prurito
Trombocitopenia Mal funcionamiento de las plaquetas Prurito Complicaciones trombóticas, arteriales o venosas Incluyen los vasos del eje esplenoportal Un 30% están asintomáticos al momento del diagnóstico 76

77
Mielofibrosis Cuadro clínico Al examen físico Esplenomegalia (85-90%)
Hepatomegalia en un 60% de los casos Palidez propia de la anemia 77

78
Mielofibrosis Cuadro clínico
La anemia habitualmente normocroma-normocítica Constituye la manifestación más frecuente (80%) Origen multifactorial Disminución en la producción medular Grado variable de eritropoyesis ineficaz Hemólisis por hiperesplenismo o de origen autoinmune Examen morfológico de la SP Policromasia, anisocitosis y poiquilocitosis, dacriocitos 78
 Origen multifactorial. Disminución en la producción medular. Grado variable de eritropoyesis ineficaz. Hemólisis por hiperesplenismo o de origen autoinmune. Examen morfológico de la SP. Policromasia, anisocitosis y poiquilocitosis, dacriocitos. 78.")
79
Mielofibrosis 79

80
Mielofibrosis Biología Síndrome leucoeritroblástico
Presencia de eritroblastos y células mieloides inmaduras en sangre periférica. Cifra de leucocitos variable Se puede observar basofilia menor frecuencia que en la LMC Trombocitopenia 25% , trombocitosis 25% Alteraciones morfológicas de las plaquetas (anisocitosis, elementos gigantes, plaquetas azules o vacualizadas) Índice de FAL se halla incrementado 80
 Índice de FAL se halla incrementado. 80.")
81
Mielofibrosis Laboratorio LDH incrementado (85%) otras alteraciones
Incremento de la FA, B12, y ácido úrico Hipocolesterolemia. Punción medular infructuosa Punción blanca debido a la gran dureza del hueso Alteraciones citogenéticas del 13q, del 20q, trisomia 1q 81

82
Mielofibrosis Anatomía patológica
La Biopsia de la MO es imprescindible para establecer el diagnóstico Es la única manera de poner de manifiesto la fibrosis medular 82

83
Mielofibrosis Diagnóstico
Existencia de fibrosis de la MO sin una causa que la justifique Esplenomegalia Síndrome leucoeritroblástico Anisopoiquilocitosis y dacriocitos Son muy frecuentes pero pueden faltar en algunos casos Diagnostico diferencial muy amplio 83

84
Mielofibrosis Evolución y pronóstico Curso crónico, caracterizado por
Anemización progresiva con consiguiente requerimiento transfusional Molestias derivadas del agrandamiento del bazo Presencia de síntomas constitucionales Supervivencia mediana 3,5 a 5 años 84

85
Preguntas?... 85

Presentaciones similares






>")












