Descargar la presentación
La descarga está en progreso. Por favor, espere

1
DEFINICIÓN: Síndrome que se caracteriza por la baja circulante de G R (Hb ó Ht)*. *Excepto anemia por hemorragia aguda. DR. MARIO GUTIERREZ ROMERO HOSPITAL GENERAL DE MEXICO,O.D. E mail: mgutierrezr_2000 @ yahoo.com.mx SINDROME ANÉMICO

2
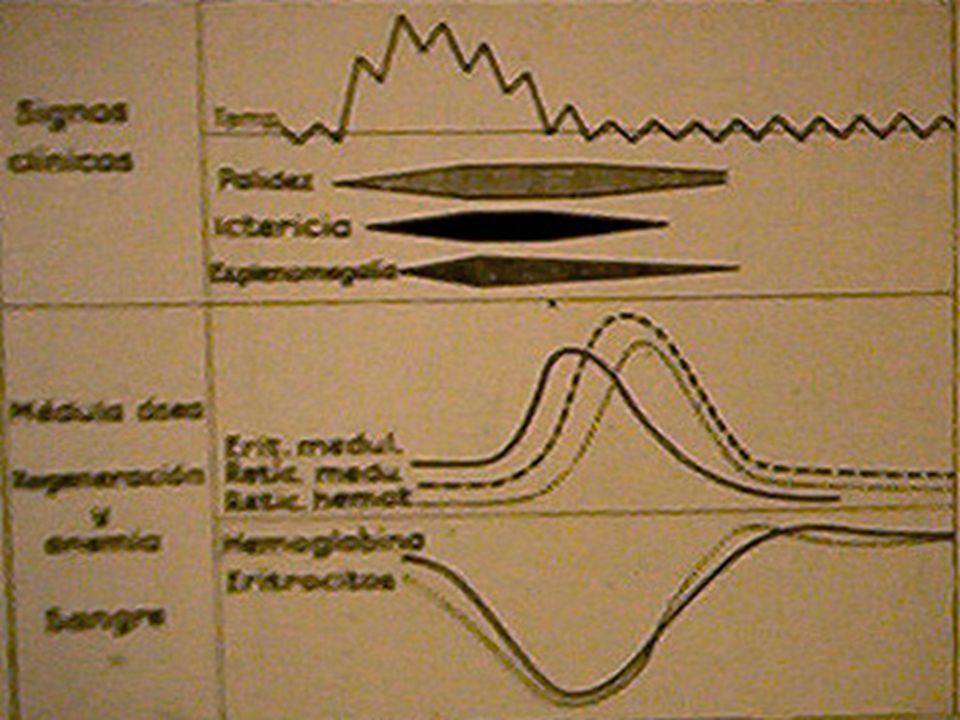
*Diferenciar ictericia y anemia, si se presentan es ANEMIA HEMOLITICA. **Otros datos específicos de anemia se verán en los capítulos correspondientes. SIGNOS Y SINTOMAS DEL SINDROME ANEMICO S. ANEMICO APARATO / SISTEMA SINTOMASSIGNOS PALIDEZ PIEL Y MUCOSAS SNC.CEFALEA, MAREO, LIPOTIMIA (ICTERICIA)*. ACUFENOS TAQUISFIGMIA. FOSFENOS. TAQUICARDIA. CARDIOVASCULARLATIDOS, PALPITACIONES, ANGOR. SOPLOS FUNCIONALES INSUFICIENCIA CARDIACA BAJA DE GR RESPIRATORIO DISNEA. TAQUIPNEA MUSCULAR ASTENIA, ADINAMIA. ESPECIFICOS DE ALGUNOS TIPOS DE ANEMIA** HIPOXIA TISULAR DIGESTIVO HIPOREXIA, DIARREAS. LENGUA LISA, MACROGLOSIA, ESPLENOMEGALIA RENALEDEMAS REPRODUCTORAMENORREA, ESTERILIDAD. PIEL Y ANEXOSUÑAS QUEBRADIZAS CAIDA DE PELO PLATONIQUIA, COILONIQUIA, PIEL SECA, PELO SECO
*. ACUFENOS TAQUISFIGMIA. FOSFENOS. TAQUICARDIA. CARDIOVASCULARLATIDOS, PALPITACIONES, ANGOR. SOPLOS FUNCIONALES INSUFICIENCIA CARDIACA BAJA DE GR RESPIRATORIO DISNEA. TAQUIPNEA MUSCULAR ASTENIA, ADINAMIA. ESPECIFICOS DE ALGUNOS TIPOS DE ANEMIA** HIPOXIA TISULAR DIGESTIVO HIPOREXIA, DIARREAS. LENGUA LISA, MACROGLOSIA, ESPLENOMEGALIA RENALEDEMAS REPRODUCTORAMENORREA, ESTERILIDAD. PIEL Y ANEXOSUÑAS QUEBRADIZAS CAIDA DE PELO PLATONIQUIA, COILONIQUIA, PIEL SECA, PELO SECO.")
3
CRITERIO DE LA ORGANIZACIÓN MUNDIAL DE LA SALUD (OMS). ANEMIA POR LABORATORIO (BH) CUANDO LA CIFRA DE Hb < 12g/dL
 CUANDO LA CIFRA DE Hb < 12g/dL.")
4
CLASIFICACIÓN DEL GRADO DE ANEMIA. GRADOHEMOGLOBINA I De lo normal a 10 g/dL II De 9.9 a 8.0 g/dL III < 8.0 g/dL

5
CLASIFICACIÓN DE LA ANEMIA DE ACUERDO A LA RESPUESTA DE LA MO REGENERATIVA:Cta. de reticulocitos > 2 % ARREGENERATIVA:Cta. de reticulocitos < 0.5 % Cuenta reticulocitos corregida por anemia Ht paciente Ht normal (0.45 L/L) =% reticulocitosX
 =% reticulocitosX.")
6
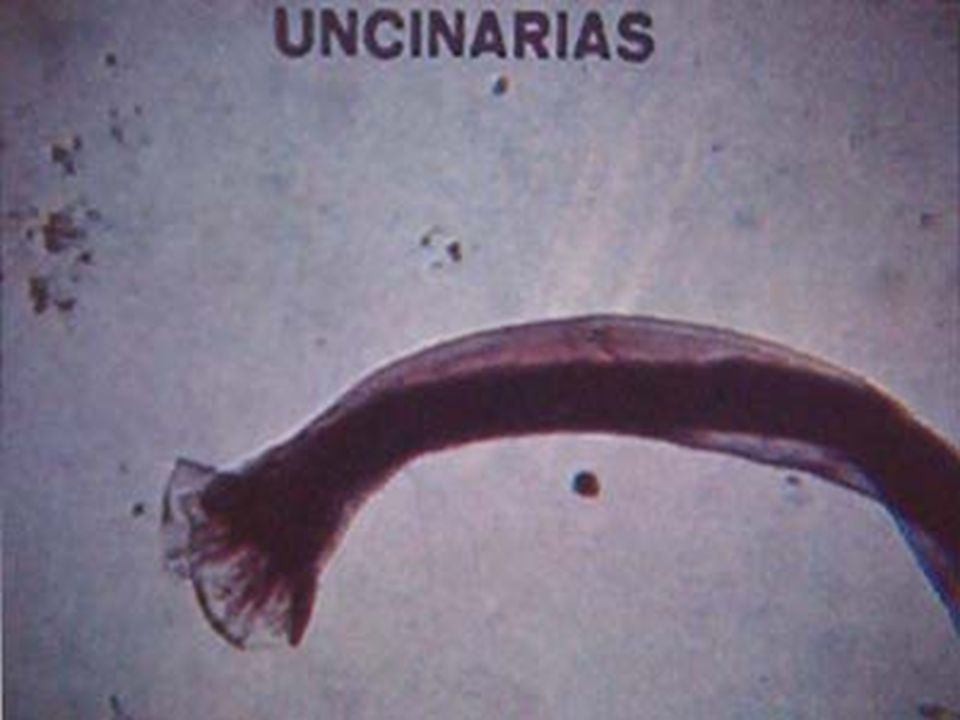
CLASIFICACIÓN ETIOPATOLOGICA DE LAS ANEMIAS. I. DEFICIENCIA DE NUTRIENTES HEMATOPOYETICOS Fe, V.B12, Ac. F Nutricional: Dieta pobre en Fe, B12 o Ac.fólico Patologías del tubo digestivo: Estómago y duodeno: Def. de Fe Estómago e Ileon: Def. de V.B 12 Yeyuno: Def. de Ac. F. II. HEMORRAGIA AGUDA Ruptura de vasos de Mediano o gran calibre III. FALLA EN LA PRODUCCIÓN DE MO S. insuficiencia medular: Leucemias, SMD, Ca. Aplasia medular: Idiopática o secundaria. Congénita Mayores requerimientos: Embarazo Hemorrágias crónicas: Uncinariasis

7
IV. AUMENTO EN LA DESTRUCCIÓN o HIPERHEMÓLISIS Daño hereditario del GR. Causas adquiridas que dañan al GR. V. MULTIFACTORIAL (CAUSA MULTIPLE) ENFERMEDADES CRONICAS*: Ca, Infección, Nefropatía, Colagenopatía, Hepatopatía, Endocrinopatía, otras. *Las más frecuentes en adultos
 ENFERMEDADES CRONICAS*: Ca, Infección, Nefropatía, Colagenopatía, Hepatopatía, Endocrinopatía, otras. *Las más frecuentes en adultos.")
8
CLASIFICACIÓN MORFOLÓGICA (fisiopatológica) DE LAS ANEMIAS. TIPO VALOR DE HCMVCM ETIOPATOGENIAFRECUENCIA NIÑOSADULTOS NORMOCITICA, NORMOCRÓMICA I N N 1.Hemorragia aguda 2.Daño en médula ósea 3.Hiperhemólisis 4.Enfermedades crónicas ** 15 %85 % II HIPOCRÓMICA NORMOCÍTICA Ó MICROCÍTICA N Deficiencia de Fe ( >99 %) Falla en utilización (<1 % ) 85 %15 % ---- III MACROCÍTICA NORMOCRÓMICA N Deficiencia de V. B12 o Ac.F. < 5 % ** La anemia de la enfermedad crónica en algún momento de su evolución puede ser de cualquier tipo. 80-100fL27-32pg <27pg >100fL
 Falla en utilización (<1 % ) 85 %15 % ---- III MACROCÍTICA NORMOCRÓMICA N Deficiencia de V. B12 o Ac.F. < 5 % ** La anemia de la enfermedad crónica en algún momento de su evolución puede ser de cualquier tipo fL27-32pg <27pg >100fL.")
9
FLUJOGRAMA DIAGNÓSTICO EN SÍNDROME ANÉMICO Paciente con datos clínicos de anemia. BH con Hb < de 12 g/dL Realizar cuenta de reticulocitos (0.5-2.0%) I.- Hipocrómica HCM < 27 pg II.- Normocítica Normocrómica VCM 80-100 fL HCM 27-32 pg III.- Macrocítica VCM > 100 fL
 I.- Hipocrómica HCM < 27 pg II.- Normocítica Normocrómica VCM fL HCM pg III.- Macrocítica VCM > 100 fL.")
10
I.- HIPOCROMICA BH: HCM < 27 pg. VCM N / (< 80 fL) Hipocromia/Microcitosis Hierro sérico. Ferritina sérica. AMO con tinción para Fe. DisminuidoAlto Anemia deficiente de Fe. Buscar la causa: Pérdida crónica de sangre. Trastorno absorción TD alto. Requerimientos aumentados. Anemia sideroacréstica: Síndrome mielodisplásico, Talasemia, Def. V. B6 (piridoxina) ( < 1 % de las causas): Paciente deficiente en Tx con Fe
 ( < 1 % de las causas): Paciente deficiente en Tx con Fe.")
11
II.- NORMOCITICA NORMOCROMICA BH: HCM Normal, VCM Normal RETICULOCITOS Altos Normales ó bajos Anemia hemolítica (ictericia) Anemia por hemorragia aguda PUNCION MO Anormal Aplasia medular Infiltración por: Leucemia, Mieloma múltiple, Linfoma. Mielofibrosis. Síndrome mielodisplásico. Normal Deficiencia temprana de Fe (?). Enfermedad crónica: renal, Ca, infección, hepática, endocrina, colagenopatia.
. Enfermedad crónica: renal, Ca, infección, hepática, endocrina, colagenopatia..")
12
III.- MACROCITICA BH: VCM > 100 fL. Macrocitosis Reticulocitos Altos Normales Pac. def. de V. B12 o folatos en tratamiento. Hemorragia crónica. Hemólisis crónica. Aspirado de MO: MEGALOBLASTOSIS SiNo Def. Ac. Fólico o V. B12 (Hacer medición en suero) Hipotiroidismo. Aplasia medular crónica. Insuficiencia renal crónica. Hepatopatía crónica.
 Hipotiroidismo. Aplasia medular crónica. Insuficiencia renal crónica. Hepatopatía crónica..")
13
REVISION DE LOS PRINCIPALES TIPOS DE ANEMIAS : 1.ANEMIAS CARENCIALES. Deficiencias de hierro, vitamina B12 y ácido fólico. 2.ANEMIA POR HEMORRAGIA AGUDA. 3.ANEMIA APLASTICA. 4.ANEMIAS HEMOLITICAS. 5. ANEMIA DE LA ENFERMEDAD CRONICA.

14
ANEMIA POR DEFICIENCIA DE HIERRO

15
GR Alimentación: Fe ++ Fe +++ 10 - 20 mg/d Fe +++ HCl Fe ++ CELULA INTESTINAL APOFERRITINA Fe++ FERRITINA Fe+++ 10 % TRANSFERRINA Fe +++ PLASMA HIGADO (DEPÓSITO) 0.5 a 1.5 g BAZO MO 3.0-3.5 g ERITB. METABOLISMO DEL Fe. Fe total = 4.0 a 5.0 g Fe otros sitios: 0.8 g Pérdidas: Pelo, uñas, epitelios: 1-2 mg / d.

16
2a2a 1a1a 4a4a 5a5a 3a3a PATOLOGÍA. Causas: PERDIDA CRONICA INGESTION ACIDEZ + ABSORCION UTILIZACION FALLA EN EL TRANSPORTE

17
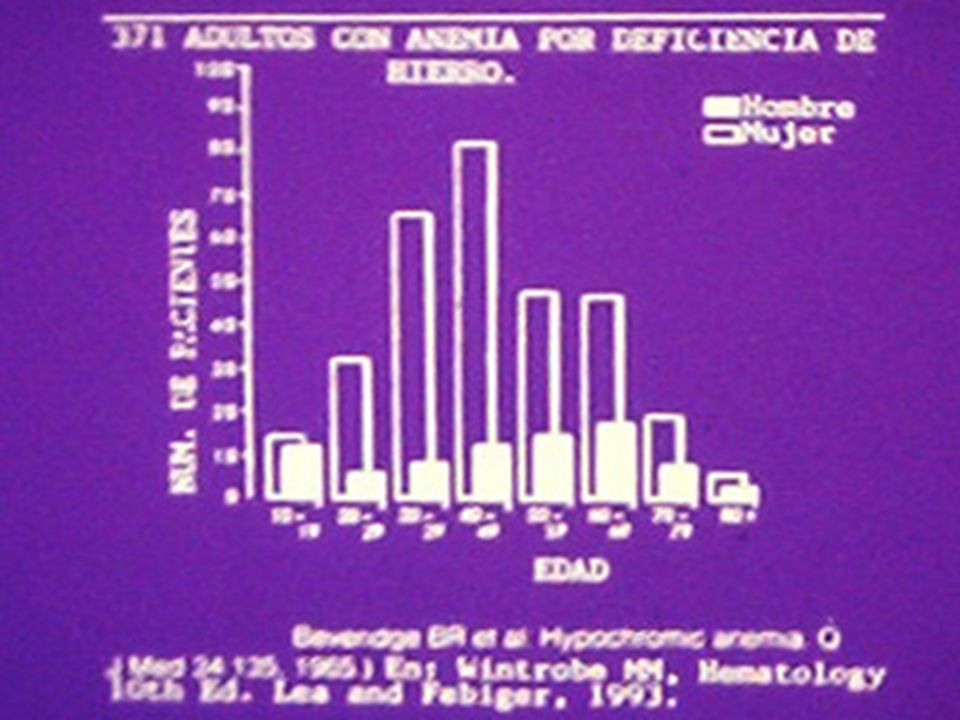
ANEMIA POR DEFICIENCIA DE Fe PRINCIPALES CAUSAS EN NUESTRO MEDIO: 1 a.- Lactantes destetados tempranamente y alimentados con leche de vaca y papillas de cereal (pobres en Fe). 2 a.- Pérdida crónica de sangre: a. Hiperpolimenorrea (morbo virgíneo), miomatosis uterina. b.- Ca T.D., pólipos o divertículos intestinales. c.- Uncinariasis (zonas cafetaleras). 3 a.- Gastritis o duodenitis crónicas, gastritis atrófica, Ca gástrico o duodenal, gastrectomía. 4 a.- Falla de la utilización en MO: SMD, leucemias, talasemias, enfermedades crónicas. 5 a.- Atransferrinemia congénita (muy rara).
, miomatosis uterina. b.- Ca T.D., pólipos o divertículos intestinales. c.- Uncinariasis (zonas cafetaleras). 3 a.- Gastritis o duodenitis crónicas, gastritis atrófica, Ca gástrico o duodenal, gastrectomía. 4 a.- Falla de la utilización en MO: SMD, leucemias, talasemias, enfermedades crónicas. 5 a.- Atransferrinemia congénita (muy rara)..")
22
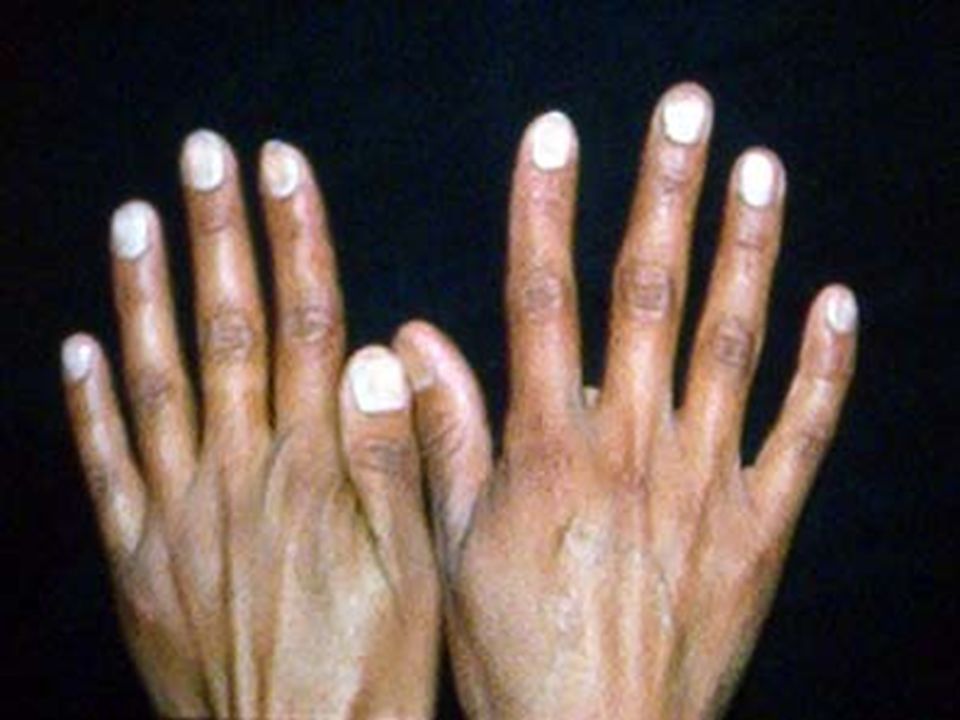
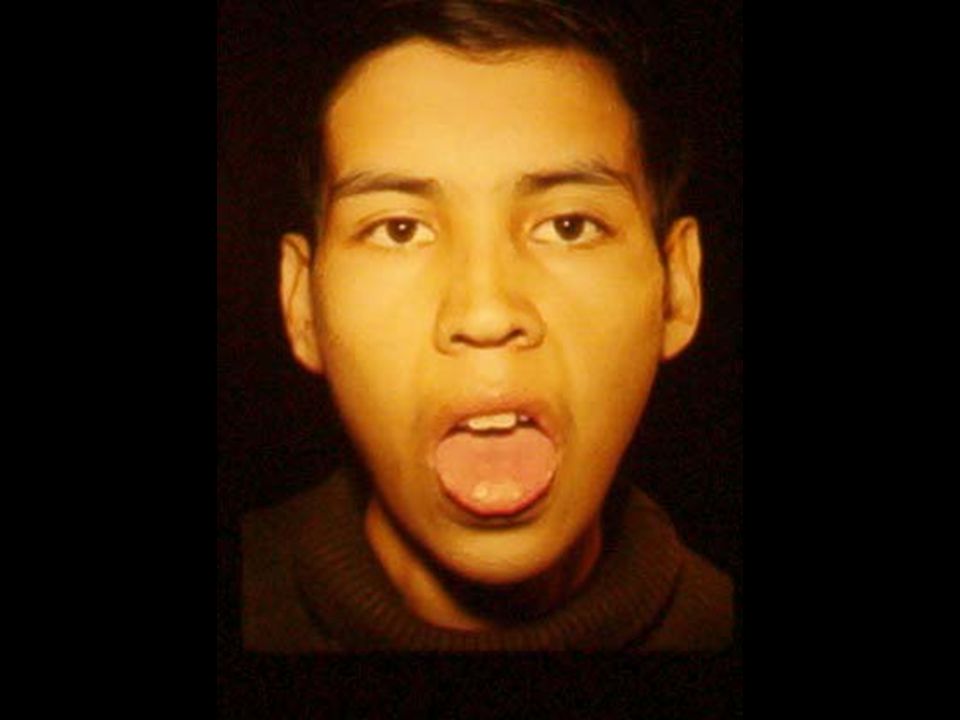
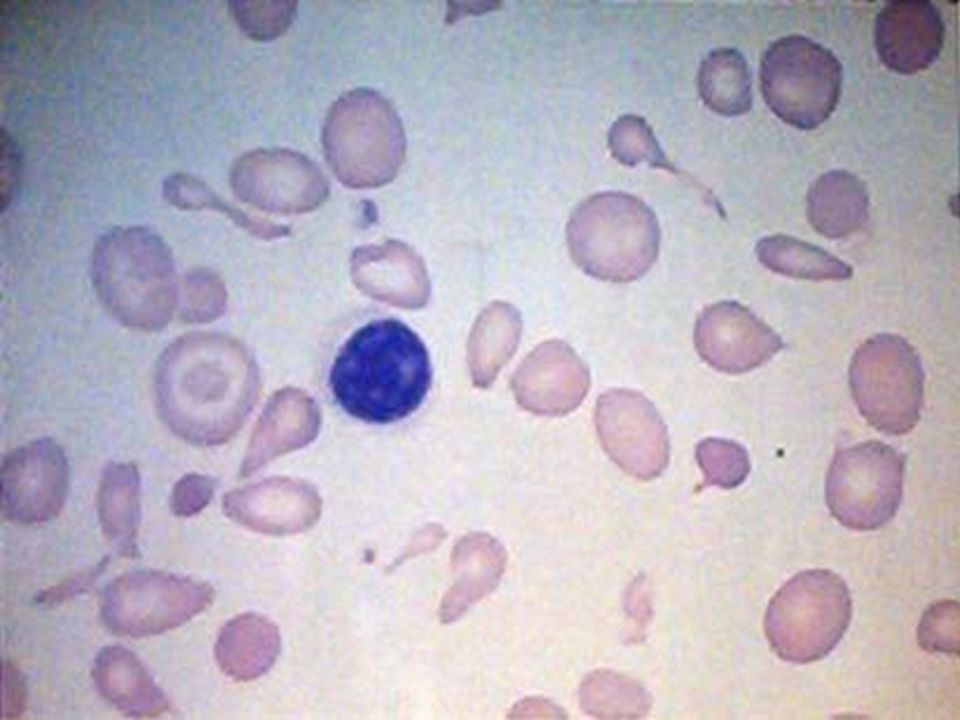
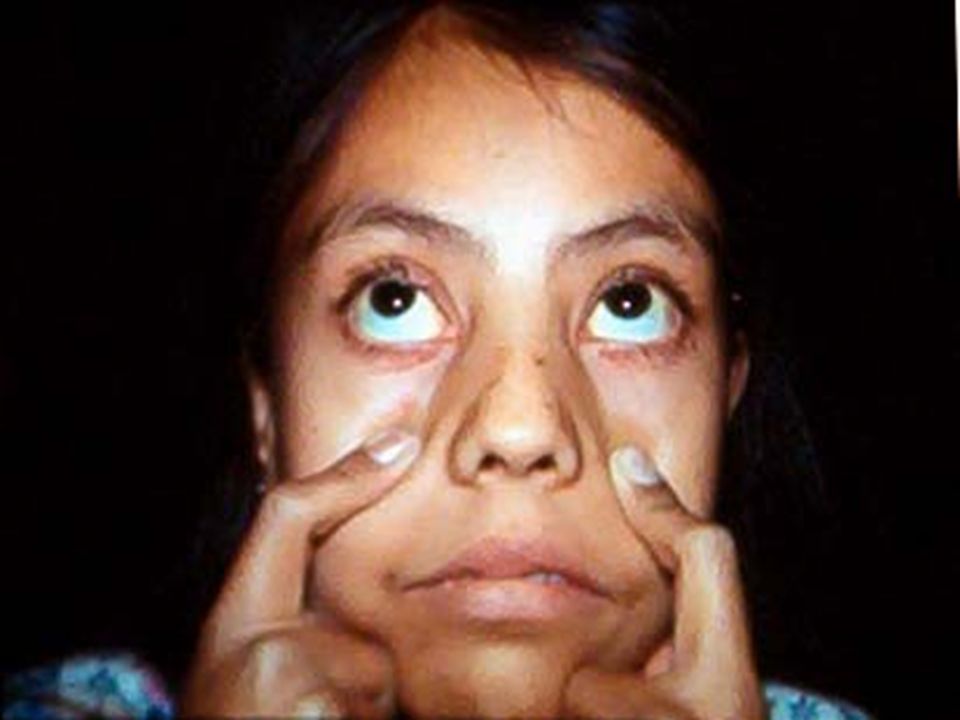
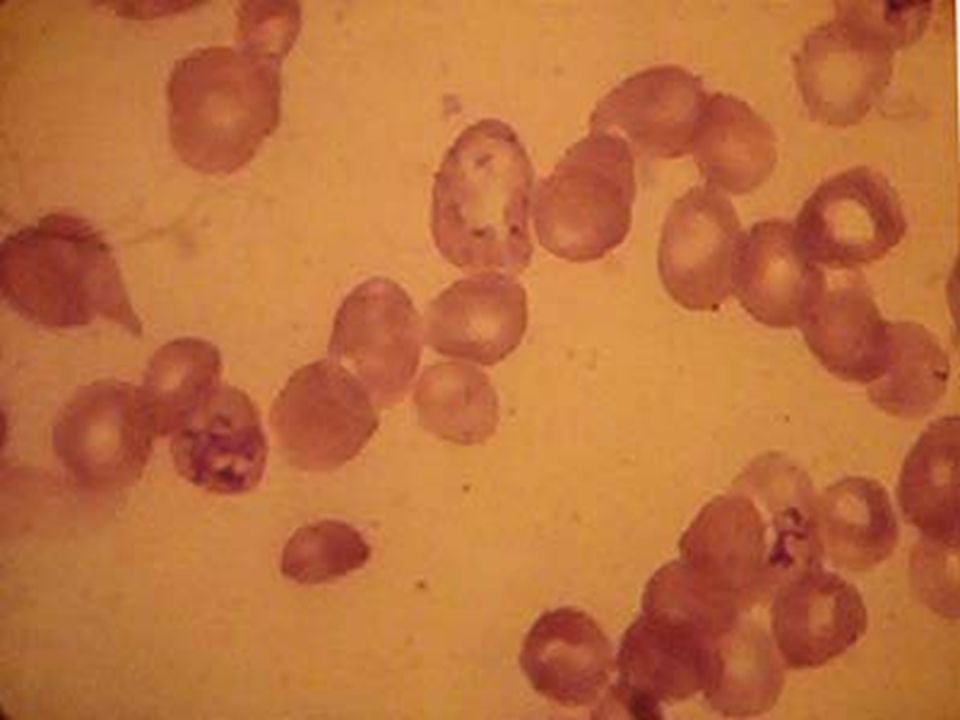
ANEMIA POR DEFICIENCIA DE Fe. Anemia mas frecuente en todo el mundo. Fe 2º metal más abunadante. Además de los datos generales de anemia: Pelo seco, quebradizo, piel seca, escamosa, uñas quebradizas, platoniquia y coiloniquia. PICA. LABORATORIO: BH: Anemia grado variable, llega a cifras extremas de 1 g/dL. Hipocrómica (HCM<27 pg) normocítica (VCM 80 – 100 fL) cuando es reciente y microcítica (VCM < 80 fL) cuando es crónica. Reticulocitos: normales ( aumentan después del tratamiento con Fe). ADE: Alto (>16%), con la curva desviada a la izquierda. Resto de las series normales, excepto trombocitosis 20-30% de los casos cuando la hemorragia es crónica. Eosinofilia en uncinariasis. Frotis: hipocromia, anisocitosis con microcitosis. MO: Reacción eritroblástica, microcitosis de la serie roja. Tinción Fe en MO: Extracelular bajo. Intracelular bajo. CUADRO CLÍNICO:
 normocítica (VCM 80 – 100 fL) cuando es reciente y microcítica (VCM < 80 fL) cuando es crónica. Reticulocitos: normales ( aumentan después del tratamiento con Fe). ADE: Alto (>16%), con la curva desviada a la izquierda. Resto de las series normales, excepto trombocitosis 20-30% de los casos cuando la hemorragia es crónica. Eosinofilia en uncinariasis. Frotis: hipocromia, anisocitosis con microcitosis. MO: Reacción eritroblástica, microcitosis de la serie roja. Tinción Fe en MO: Extracelular bajo. Intracelular bajo. CUADRO CLÍNICO:.")
23
ETAPAS DE LA HIPOFERREMIA I.- Hipoferremia (SUBCLINICA). II.- Hipoferrenia con deficiente eritropoyesis (SUBCLINICA). Ferritina sérica Fe en MO Ferritina sérica Fe en MO ADE HCM / VCM ANEMIA Capacidad de fijación Fe sérico e Ind. Saturación Ferritina sérica Fe en MO Fe sérico e Ind. Saturación C. de fijación Fe Sérico: 75-150 g/dL Cap. de fijación: 250-400 g/dL Indice de saturación: 20-40% Ferritina sérica: 35-100 g/L Protoporfirina eritrocítica libre (PEL) : 32-62 g/dL de GR. PEL (> 250 g/dL) Valores normales: III.- ANEMIA DEFICIENTE EN Fe.
. Ferritina sérica Fe en MO Ferritina sérica Fe en MO ADE HCM / VCM ANEMIA Capacidad de fijación Fe sérico e Ind. Saturación Ferritina sérica Fe en MO Fe sérico e Ind. Saturación C. de fijación Fe Sérico: g/dL Cap. de fijación: g/dL Indice de saturación: 20-40% Ferritina sérica: g/L Protoporfirina eritrocítica libre (PEL) : g/dL de GR. PEL (> 250 g/dL) Valores normales: III.- ANEMIA DEFICIENTE EN Fe..")
24
TRATAMIENTO DE LA ANEMIA POR DEFICIENCIA DE HIERRO. HIERRO ORAL: Grageas con 60 mg de hierro elemental. (2 x 3 d.) Cápsulas de liberación prolongada 100 mg. (1 d.) Jarabe con 50 mg por c/5ml. Gotas pediátricas con 2.5 mg por c/gota. HIERRO PARENTERAL: INTRAMUSCULAR Ampolletas de 2 ml con 100 mg. Aplicarlo con la técnica en Z c/3er. Día. INTRAVENOSO Ampolletas de 5 ml con 100 mg. Riesgo: choque anafiláctico.
 Cápsulas de liberación prolongada 100 mg. (1 d.) Jarabe con 50 mg por c/5ml. Gotas pediátricas con 2.5 mg por c/gota. HIERRO PARENTERAL: INTRAMUSCULAR Ampolletas de 2 ml con 100 mg. Aplicarlo con la técnica en Z c/3er. Día. INTRAVENOSO Ampolletas de 5 ml con 100 mg. Riesgo: choque anafiláctico..")
25
ANEMIA POR DEFICIENCIA DE VITAMINA B12 Y ÁCIDO FÓLICO

26
METABOLISMO DE VITAMINA B12 Y ÁCIDO FÓLICO. Hígado 5 – 10 mg Hígado 1,000 –3,000 g Depósito: Cantidad: Yeyuno Ileon. Requiere F.I. Sitio: 5 mg/d 1 mg/d 5 g/d 1 g/d Ingesta día: Absorción: Vegetal/Animal AnimalOrigen: Vitamina B12Acido fólico

27
Vitamina B12 Nucleósidos PO 2 H 2 Nucleótidos Proteínas Nucleoproteinas Ribosa o desoxirribosa Ácido fólico Ácido folínico Síntesis de las Purinas y pirimidinas + + +

29
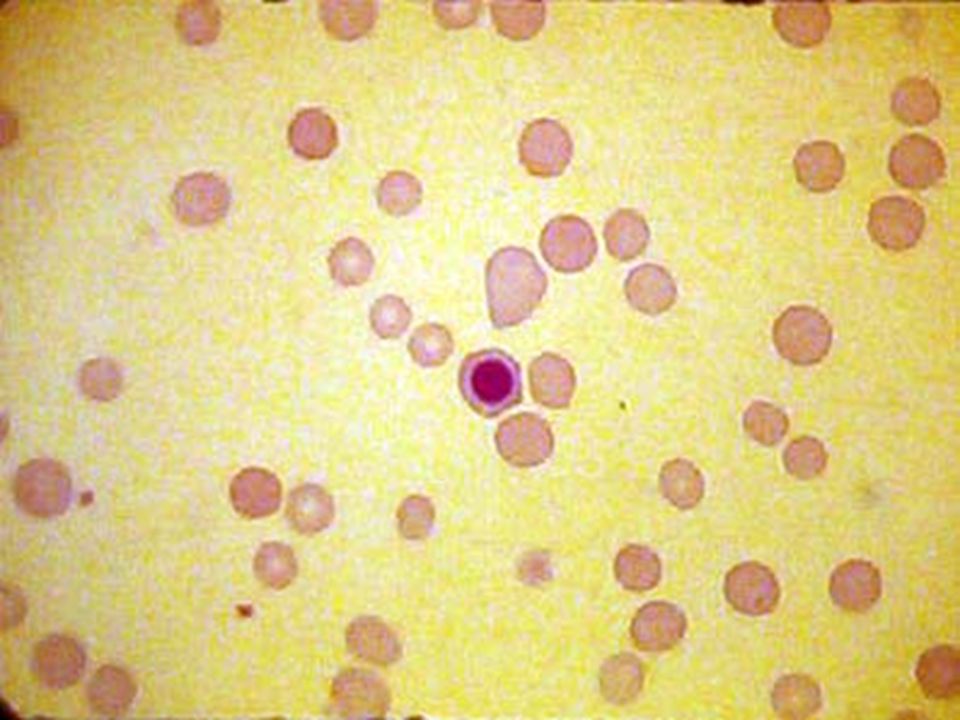
ANEMIA MACROCITICA (DEF. VIT. B12 / AC. FÓLICO) 1.Pobre ingesta: Carnes (B12), Vegetales (Ac. Fólico). 2.Trastornos gástricos: Gastritis atrófica o alcohólica, gastrectomía, (No secreción de Factor intrínseco: ANEMIA PERNICIOSA). 3. Síndromes de mala absorción intestinal: Sprue, Enfermedad celiaca. 4. Interferencia por medicamentos: TMT/SMTX, Methotrexate. 5. Mayores requerimientos: Embarazos. 6. Pérdidas anormales: Diarreas crónicas. Síndrome:Gloso-gástro-entero-anémico. Vit.B12:Alteraciones neurológicas. Palidez terrosa, macroglosia rojo magenta, papilas atróficas. Diarreas. Vitamina B12: Paresias, parestesias, hipoestesia, pérdida de la sensibilidad profunda (prueba del diapasón), signo de Romberg +, ataxia. CAUSAS: CUADRO CLÍNICO: LABORATORIO: BH: Pancitopenia. GR: VCM >120 fL, macroovalocitos, poiquilocitos, GB: neutrófilos polisegmentados. MO: Megaloblastosis. DHL elevada.
. 2.Trastornos gástricos: Gastritis atrófica o alcohólica, gastrectomía, (No secreción de Factor intrínseco: ANEMIA PERNICIOSA). 3. Síndromes de mala absorción intestinal: Sprue, Enfermedad celiaca. 4. Interferencia por medicamentos: TMT/SMTX, Methotrexate. 5. Mayores requerimientos: Embarazos. 6. Pérdidas anormales: Diarreas crónicas. Síndrome:Gloso-gástro-entero-anémico. Vit.B12:Alteraciones neurológicas. Palidez terrosa, macroglosia rojo magenta, papilas atróficas. Diarreas. Vitamina B12: Paresias, parestesias, hipoestesia, pérdida de la sensibilidad profunda (prueba del diapasón), signo de Romberg +, ataxia. CAUSAS: CUADRO CLÍNICO: LABORATORIO: BH: Pancitopenia. GR: VCM >120 fL, macroovalocitos, poiquilocitos, GB: neutrófilos polisegmentados. MO: Megaloblastosis. DHL elevada..")
32
NIVELES SÉRICOS DE VIT. B12 Y ÁCIDO FÓLICO. Ácido fólico: 6 – 20 ng/ml. Deficiencia: < 4 ng/ml. -PRUEBA DE SCHILLING: 1. Dar cápsula Vit.B12 - Co 60.. Saturar IM 1000 g Vit.B12. A las 24 hs en orina radioactividad 12 –30 % (normal). En anemia perniciosa < 4%. 2.Repetir + una cápsula de Factor intrínseco de cerdo v.o. La radioactividad en orina a las 24 hs > 12%. -BIOPSIA DE MUCOSA GÁSTRICA: Gastritis atrófica. Buscar por técnicas de Inmunohistoquímica: Ac vs F.I. VITAMINA B12: 150 – 800 pg/ml. Deficiencia: < 80 pg/ml. ANEMIA PERNICIOSA
. En anemia perniciosa < 4%. 2.Repetir + una cápsula de Factor intrínseco de cerdo v.o. La radioactividad en orina a las 24 hs > 12%. -BIOPSIA DE MUCOSA GÁSTRICA: Gastritis atrófica. Buscar por técnicas de Inmunohistoquímica: Ac vs F.I. VITAMINA B12: 150 – 800 pg/ml. Deficiencia: < 80 pg/ml. ANEMIA PERNICIOSA.")
33
TRATAMIENTO DEFICIENCIA DE VITAMINA B12 Ampolletas de 100 mcg, por vía I.M. 1er. Semana una diaria. 2a, 3a y 4a Semana una c/3er. Día. 5a. Semana hasta los 3 meses una c/semana. Valorar cada 6 meses. TRATAMIENTO DEFICIENCIA DE ACIDO FOLICO Tabletas de 5 mg. Tomar 3 diarias por un mes o más. Para vía I.M. hay preparados de ácido folínico.

34
ANEMIA POR HEMORRAGIA AGUDA

35
En los adultos, después de la anemia por enfermedad crónica la anemia por hemorragia aguda es 2º lugar en frecuencia. Las causas de pérdida aguda de sangre se clasifican en: 1.INTERNAS. P. ej: Ruptura de aneurisma aórtico. Embarazo extrauterino. Estallamiento de víscera sólida. 2.EXTERNAS. P. ej: Traumatismos Ulcera gástrica. Varices esofágicas. Aborto. Posparto. Quirúrgicas, etc.

37
FISIOLOGÍA DE LA MICROCIRCULACIÓN Arteria > 50 µm Esfinter precapilar Cabo arterial del capilar 5 µm Cabo venoso del capilar 9 µm Vénula colectora Capilar verdadero Metarteriola 10 – 15 µm Arteriola 20 – 50 µm Ganong WF: Fisoterapia médica Ed. Manual moderno, México, 1984

39
ANEMIA POR HEMORRAGIA AGUDA PÉRDIDAMANIFESTACIONES CLÍNICAS 10 % Ocasionalmente manifestaciones vagales. 20 % Decúbito: asintomático o sensación de frío o ansiedad. De pié: Mareos, acufenos, fosfenos, ambliopía, debilidad, sudoración, nausea, cefalea, taquicardia, taquisfigmia, polipnea, respiración superficial, desmayo, hipotensión, algunos cambios en TA y PVC. 30 % Permanece en decúbito presentando los datos anteriores. Se agrega palidez, sed intensa, inquietud, confusión, sudor frío, pulso filiforme, hipotermia, oliguria, tono cardiaco débil, soplos cardiacos holosistolicos, retardo del llenado capilar, venas colapsadas, baja TA y PVC. 40 % Indiferencia, cianosis, anuria, colapso vascular, estupor, respiración tipo Kussmaul, choque hipovolémico. 50 % Choque hipovolémico, coma y muerte.

40
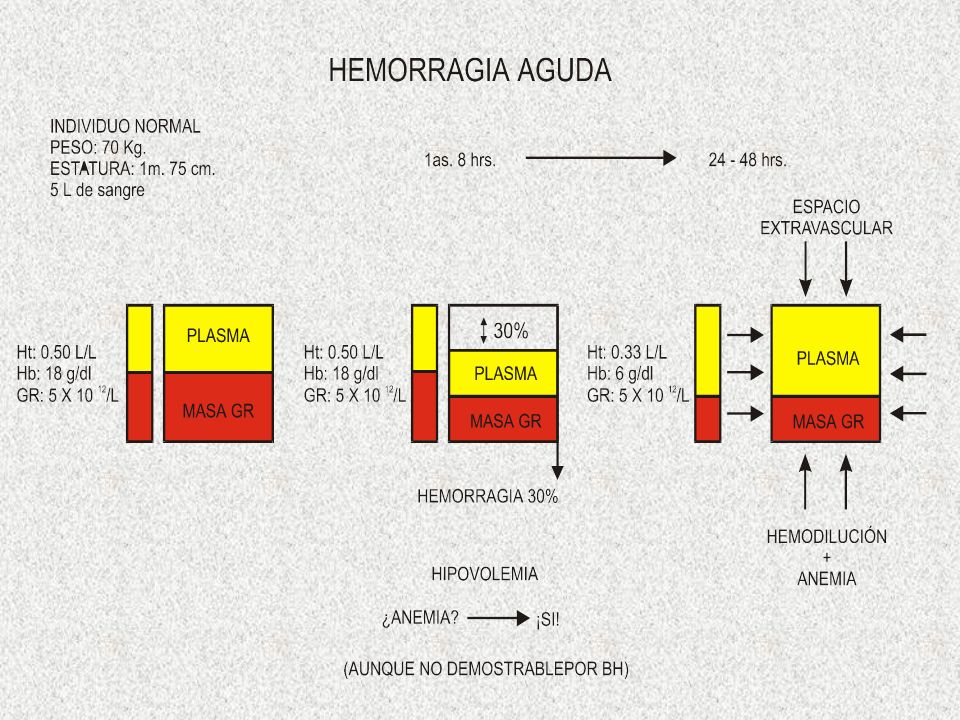
ANEMIA POR HEMORRAGIA AGUDA. Se observan 2 etapas: A. HIPOVOLEMIA. B. HEMODILUCIÓN. B. Esperar reposición espontánea del volumen plasmático es tardado*, se requiere de urgencia la reposición intravenosa de líquidos. Posteriormente por datos clínicos y BH se puede determinar el grado de anemia. La respuesta reticulocitaria es en 6-12 h. La MO es hiperplásica 2 a 5 - 10 días. Leucocitosis 10-30x10 9 /L y trombocitosis 1,000x10 9 /L dura 5 días. Recuperación total 8-9 semanas. A.Etapa mas crítica. choque hipovolemico puede causar la muerte si la pérdida es > 40%. Pérdidas 20% requieren 20–60 h para restaurar el volumen plasmático*. Hemorrágia en 24 h del 50% el organismo la tolera. Si es brusca en 30 min se pone en riesgo la vida. En esta etapa determinaciones de GR, Hb y Ht no son útiles.

41
FISIOPATOGENIA DE LA ANEMIA SEVERA Los valores mínimos de Hemoglobina requeridos para oxigenar la sangre son 5 g /dL, de acuerdo a los mecanismos de adaptación de un individuo normal. Carson y col. 125 pacientes testigos de Jehová con anemia que requerían intervención quirúrgica: Hb 8 g/dL y pérdida sanguínea < 500 ml: mortalidad nula. Hb < 8 g/dL y pérdida incluso < 500 ml: falleció el 38%*. *Se vio que los pacientes cursaban con factores de riesgo como: cardiopatía, alteraciones del potasio, diabetes, prótesis valvulares, nefropatía o hepatopatía. En 8,787 cirugías de cadera Hb 8 g/dL no afectó mortalidad. En UCI 69 pacientes con disfunción orgánica múltiple: Hb 9 vs Hb 11 g/dL, no se afectaba la mortalidad. Se concluye que Hb preoperatoria es predictor de mortalidad. En cirugía mayor: mínimo de Hb requerida 8 g /dL.

42
ANEMIA APLÁSTICA

43
ANEMIA APLASTICA ( AA ). 1888 Paul Ehrlich publicó el primer caso. Hipoplasia medular. An. Arregenerativa. Síndromes insuficiencia medular. ¿ Enfermedad autoinmune celular ? Linfocitos T citotóxicos CD8 vs células progenitoras mieloides CD34. CLASIFICACIÓN. Congénita: Anemia de Fanconi y Aplasia pura de serie roja. Adquirida : Primaria o Idiopática. Secundaria:-Agentes físicos: Radiación ionizante. -Químicos: Benceno, insecticidas, medicamentos -Infecciones: Hepatitis C. IDIOPATICA: Etiología NO SE CONOCE.

44
ANEMIA APLÁSTICA CAUSAS (Estudio de 122 casos) DDT42 % Thiner 8 % DDT + Thiner 5% DDT + Pirazolona 1 % Tolbutamida 1 % Sin antecedente43 % Santiago A.I. Anemia aplástica. Monografía Num 6 Soc. Med. H.G. México, 1981.

45
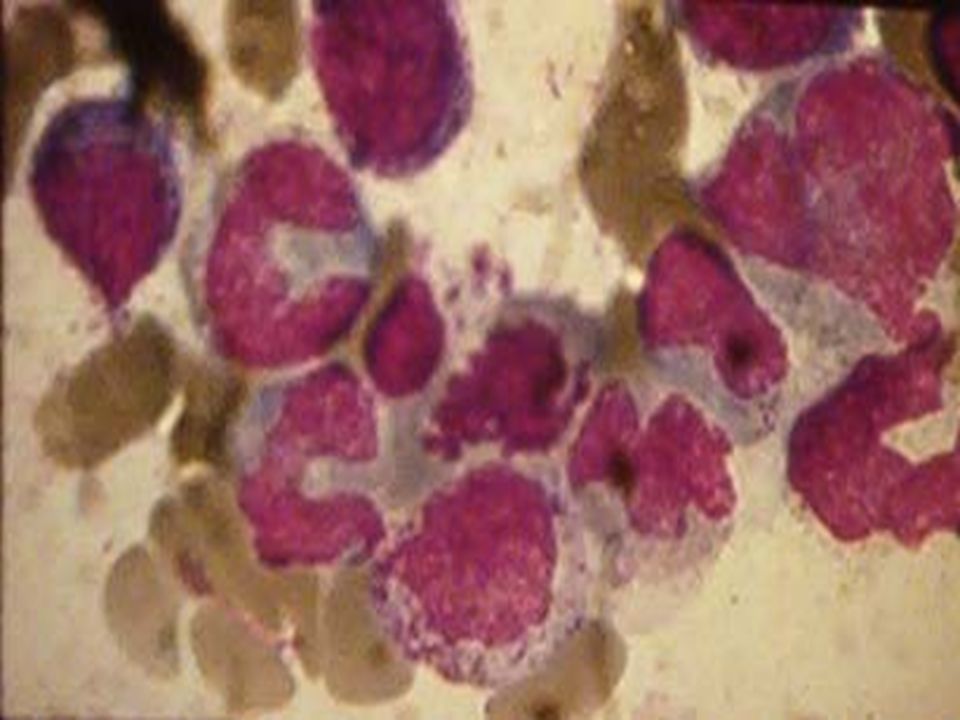
Predomina anemia crónica + S. purpúrico + S. infeccioso. De inicio aparentemente sano. No hay adeno-visceromegalias, no dolor óseo. Evoluciona a estado ± grave 3-6 meses o mas. BH: Pancitopenia, predomina anemia NN. Reticulocitos bajos (<0.2%). Leucopenia con neutropenia (< 500 L). Plaquetas < 20,000 L. Estos son criterios para AA severa. Si no los tiene es moderada. AMO: hipocelularidad marcada. BMO: relación MO roja <10% MO amarilla >90%. (normal en joven 50:50 y en adulto 30:70%). Linfocitos T CD4/CD8: invertida (normal 2:1). HbF > 3%. Dx. Diferencial: LA, SMD, HPN, Mielofibrosis. AA Severa: GAL + Ciclosporina vs TRANSPLANTE DE MO. Moderada: Anabólicos, EPO. ANEMIA APLÁSTICA CUADRO CLÍNICO: LABORATORIO: TRATAMIENTO:
. Leucopenia con neutropenia (< 500 L). Plaquetas < 20,000 L. Estos son criterios para AA severa. Si no los tiene es moderada. AMO: hipocelularidad marcada. BMO: relación MO roja <10% MO amarilla >90%. (normal en joven 50:50 y en adulto 30:70%). Linfocitos T CD4/CD8: invertida (normal 2:1). HbF > 3%. Dx. Diferencial: LA, SMD, HPN, Mielofibrosis. AA Severa: GAL + Ciclosporina vs TRANSPLANTE DE MO. Moderada: Anabólicos, EPO. ANEMIA APLÁSTICA CUADRO CLÍNICO: LABORATORIO: TRATAMIENTO:.")
46
ANEMIA APLASTICA Estudio de 122 casos Manifestaciones clínicas: S. Anémico S. Purpúrico Infección Hematología, H.G.M. 122 118 91 100 % 97 % 75 %

52
ANEMIAS HEMOLÍTICAS

53
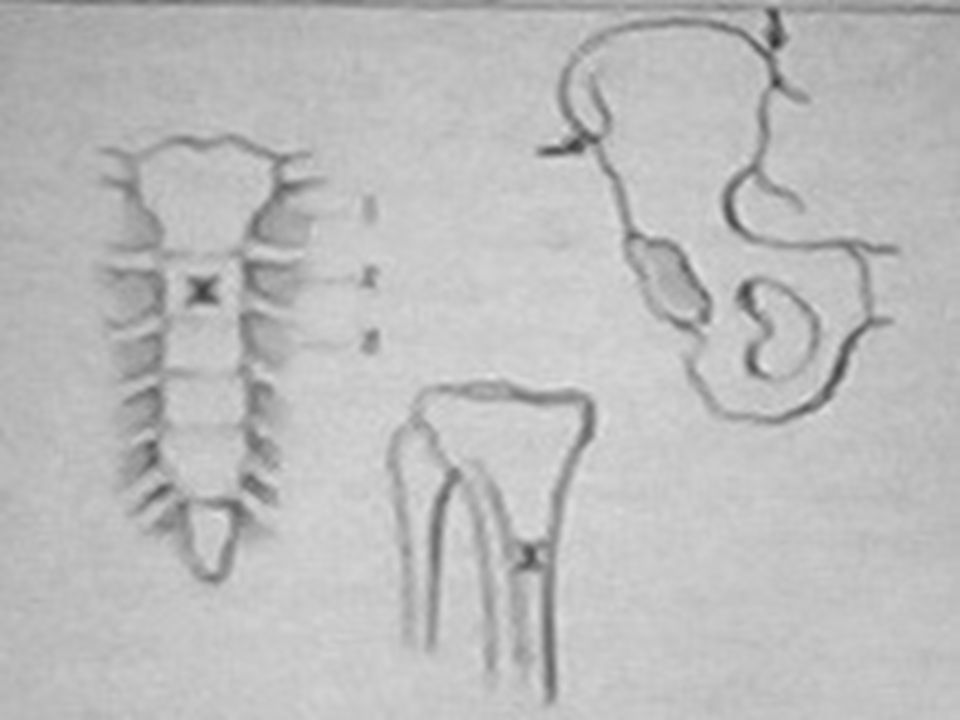
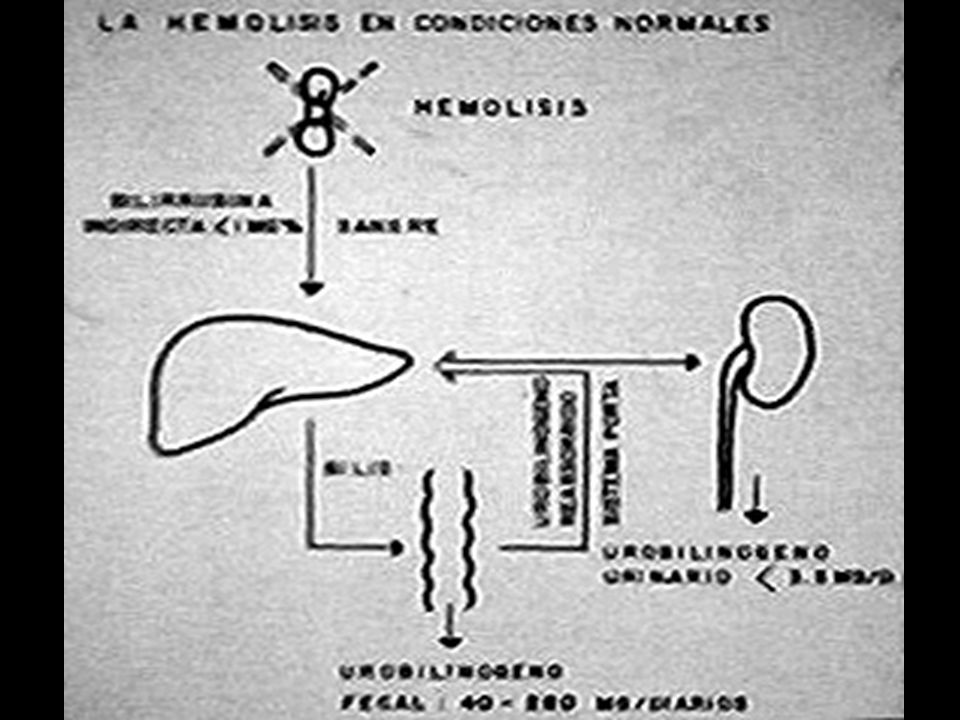
DESTRUCCION NORMAL DE GLOBULOS ROJOS TIPOS DE HEMOLISIS: I. FAGOCITOSIS : Los GR opsonizados o dañados son captados por los macrófagos del bazo en donde se rompen. HEMO: Tetrapirrol lineal +Fe (BILIVERDINA). Desprende Fe y pasa a: Plasma donde se une a la albúmina: Bilirrubina Indirecta (1mg/dL). Llega al HIGADO liga Ac. Glucurónico: Bilirrubina Directa forma bilis. Por oxidación y reducción en intestino: UROBILINOGENO FECAL. Se elimina 50% por heces: ESTERCOBILINA 40 a 250 mg/d. Se reabsorbe 50%, una parte se elimina por orina: UROBILINOGENO URINARIO 0.1 a 5 mg/d, el resto por hígado y vías biliares. Ictericia ( BI > 1 mg/dL). Heces hipercólicas o pleyocromia. Orina hipercromúrica (urobilinógeno > 5 mg). Anemia. Reticulocitosis. GR libera: H 2 O + HEMO + GLOBINA. DATOS DE HIPERHEMOLISIS:
. Desprende Fe y pasa a: Plasma donde se une a la albúmina: Bilirrubina Indirecta (1mg/dL). Llega al HIGADO liga Ac. Glucurónico: Bilirrubina Directa forma bilis. Por oxidación y reducción en intestino: UROBILINOGENO FECAL. Se elimina 50% por heces: ESTERCOBILINA 40 a 250 mg/d. Se reabsorbe 50%, una parte se elimina por orina: UROBILINOGENO URINARIO 0.1 a 5 mg/d, el resto por hígado y vías biliares. Ictericia ( BI > 1 mg/dL). Heces hipercólicas o pleyocromia. Orina hipercromúrica (urobilinógeno > 5 mg). Anemia. Reticulocitosis. GR libera: H 2 O + HEMO + GLOBINA. DATOS DE HIPERHEMOLISIS:.")
58
II. HEMOLISIS INTRAVASCULAR. El 10% de GR se rompen en la circulación liberándose la Hb, ésta es captada por las HAPTOGLOBINAS del plasma y llevada al hígado para seguir su proceso metabólico. Anemia, dolor lumbar, fiebre, hipotensión, choque, hemoglobinuria (Hb:PM 65,000 D, se filtra por el glomérulo), Haptoglobinas en plasma: 0 (completamente saturadas), en casos extremos metalbúmina y albúmina transportan Hb. La Hb libre es tóxica para el túbulo renal. *Este tipo de hiperhemólisis es poco frecuente. DATOS DE HIPERHEMOLISIS*: Valores normales de HAPTOGLOBINAS en plasma: 0.33 - 2.13 g/dL
, Haptoglobinas en plasma: 0 (completamente saturadas), en casos extremos metalbúmina y albúmina transportan Hb. La Hb libre es tóxica para el túbulo renal. *Este tipo de hiperhemólisis es poco frecuente. DATOS DE HIPERHEMOLISIS*: Valores normales de HAPTOGLOBINAS en plasma: g/dL.")
59
ANEMIAS HEMOLÍTICAS I.- DEFECTOS PROPIOS DEL GR. 1.- HEREDITARIOS. A.- Microesferocitosis hereditaria. B.- Drepanocitosis (Hemoglobinopatía S). C.- Talasemias. D.- Deficiencia de PC y G6PD 2.- ADQUIRIDO. Hemoglobinuria paroxística nocturna II.- CAUSAS EXTRACORPUSCULARES A.- Mecánica (Microangiopática). B.- Hiperesplenismo. C.- Autoinmune primaria. D.- Autoinmune secundaria. E.- Isoinmunes.
. C.- Talasemias. D.- Deficiencia de PC y G6PD 2.- ADQUIRIDO. Hemoglobinuria paroxística nocturna II.- CAUSAS EXTRACORPUSCULARES A.- Mecánica (Microangiopática). B.- Hiperesplenismo. C.- Autoinmune primaria. D.- Autoinmune secundaria. E.- Isoinmunes..")
60
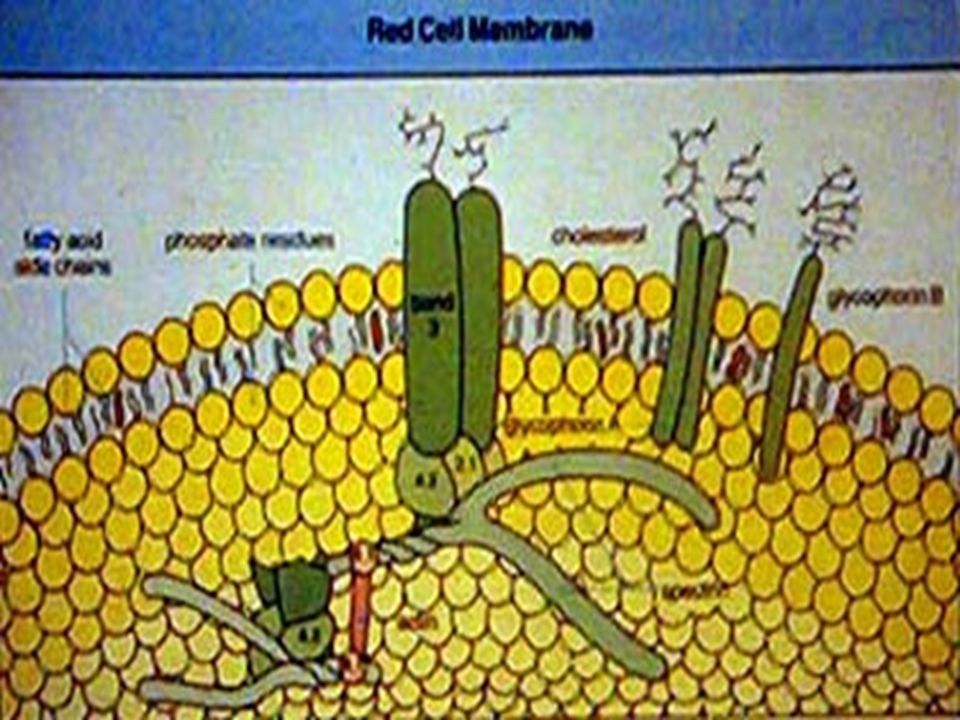
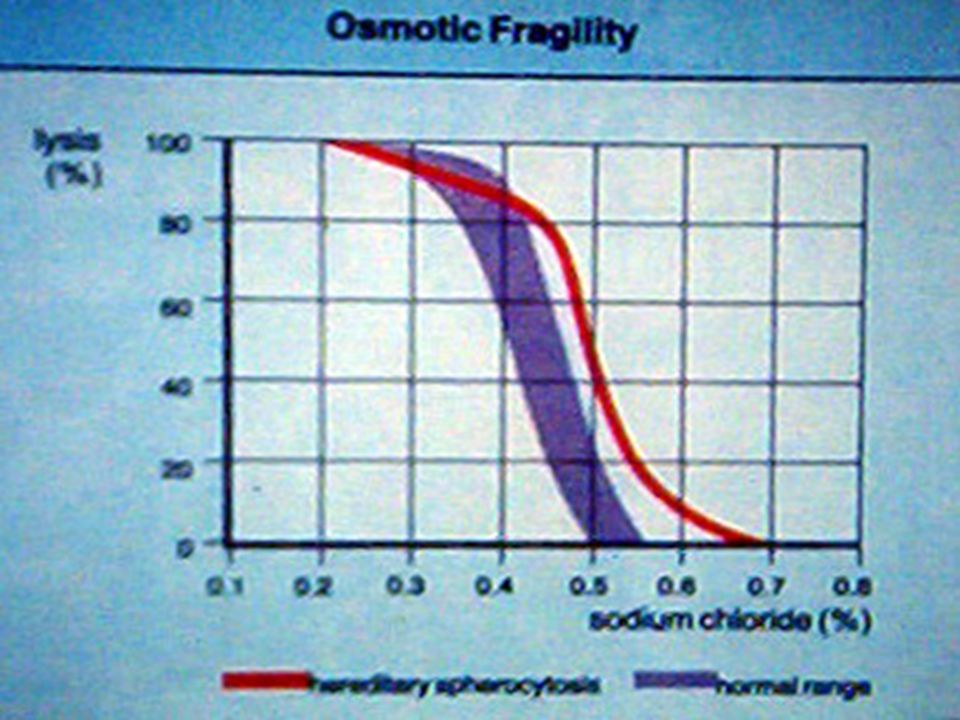
A. MICROESFEROCITOSIS HEREDITARIA. Se hereda con carácter dominante autosómico. Los microesferocitos circulan mas lentamente por los sinusoides esplénicos (<4µ de diámetro) y son fácilmente fagocitados. En procesos febriles o de hipermetabolismo sobreviene la crisis hemolítica: Anemia, ictericia, hipercromuria e hipercolia. BH: Anemia, reticulocitosis, presencia de normoblastos y microesferocitos. BI > 1 mg/dL. Urobilinógeno U. > 5 mg/d. Prueba diagnóstica: FRAGILIDAD OSMOTICA DE LOS GR A LAS SOLUCIONES SALINAS HIPOTONICAS. La hemólisis es más temprana a cambios de concentración de NaCl comparada con los GR normales. Prueba similar con glicerol acidificado. Tratamiento: Sintomático y posteriormente esplenectomía. Defecto del GR: Deficiencia de espectrina y en algunos casos de ankirina que producen GR de forma esférica.
 y son fácilmente fagocitados. En procesos febriles o de hipermetabolismo sobreviene la crisis hemolítica: Anemia, ictericia, hipercromuria e hipercolia. BH: Anemia, reticulocitosis, presencia de normoblastos y microesferocitos. BI > 1 mg/dL. Urobilinógeno U. > 5 mg/d. Prueba diagnóstica: FRAGILIDAD OSMOTICA DE LOS GR A LAS SOLUCIONES SALINAS HIPOTONICAS. La hemólisis es más temprana a cambios de concentración de NaCl comparada con los GR normales. Prueba similar con glicerol acidificado. Tratamiento: Sintomático y posteriormente esplenectomía. Defecto del GR: Deficiencia de espectrina y en algunos casos de ankirina que producen GR de forma esférica..")
64
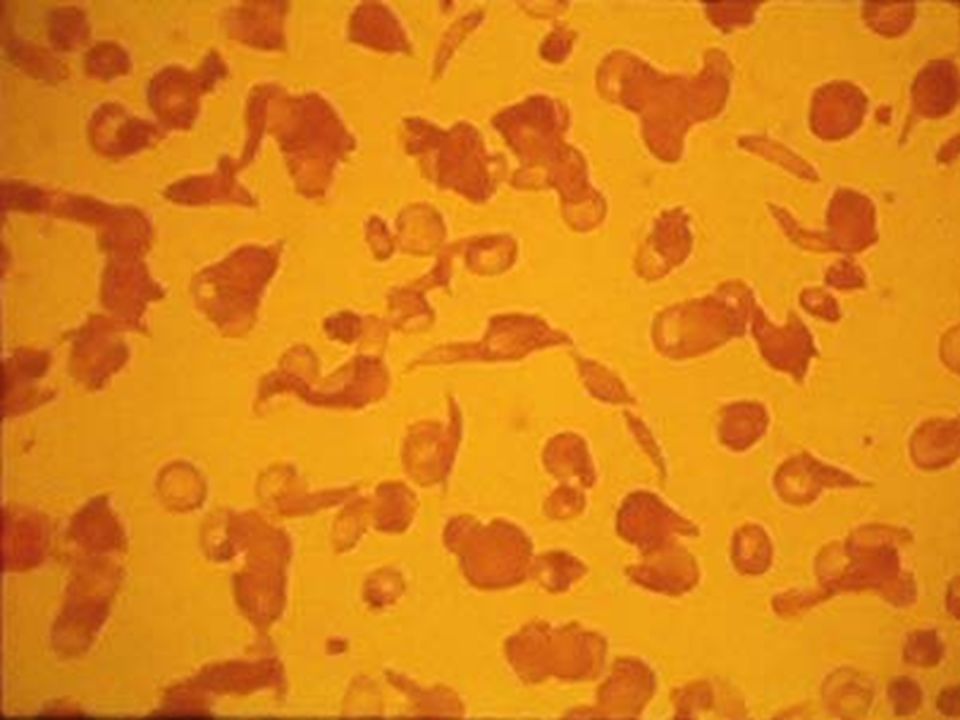
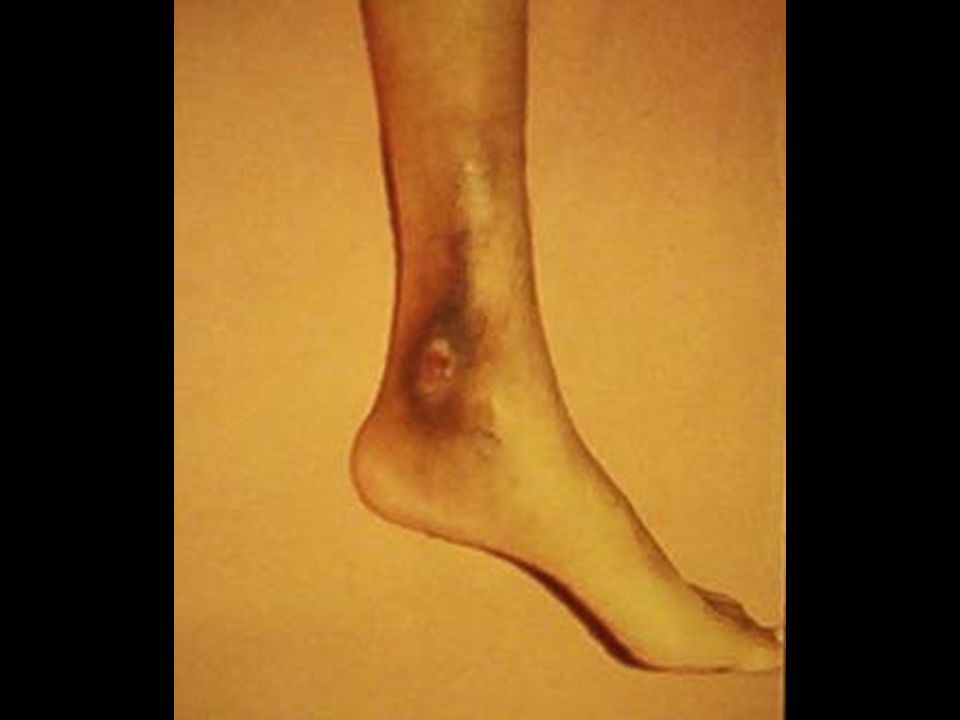
B. DREPANOCITOSIS ó ANEMIA DE CELULAS EN HOZ ó FALCIFORMES ó Hb S ó AFRICANA Herencia: Autosómica codominante. Los heterocigotos AS son portadores. Los homocigotos SS tienen la enfermedad. Defecto del GR: Es una hemoglobinopatía. En la globina cadena hay cambio en el 6º aminoácido: Acido Glutámico por valina. Este cuando baja la tensión de O 2 se enfrenta la valina del aminoácido 1º haciendo puentes de unión débiles que polimerizan la Hb formando drepanocitos. Si sube la tensión de O 2 el GR regresa a lo normal. Clínica: El paciente es normal. Cuando hay patologías que bajen la tensión de O 2 dan la crisis hemolítica. A su vez los drepanocitos producen microinfartos capilares en todos los tejidos (incluyendo pulmón). Habitualmente muestran úlceras bimaleolares. Prueba diagnóstica: Electroforesis de Hb: Portador AS. Paciente SS. Tratamiento. Sintomático: Transfusiones, Hydroxyurea, cianatos. Curativo: TMO.
. Habitualmente muestran úlceras bimaleolares. Prueba diagnóstica: Electroforesis de Hb: Portador AS. Paciente SS. Tratamiento. Sintomático: Transfusiones, Hydroxyurea, cianatos. Curativo: TMO..")
69
C. TALASEMIA ó ANEMIA MEDITERRANEA ó de COOLEY Herencia: Autosómica dominante. Clínica: Tratamiento: Sintomático. Curativo TMO. Alteración del GR: Disminución en producción de cadenas Hb:,,, y combinaciones. Las comunes y. Homocigotas (talasemia mayor). Heterocigotas (talasemia menor). Talasemia 4 variantes: Talasemia menor. Moderada anemia hemolítica y sideroblastica, hipocromia y microcitosis. Hb 10-13 g/dL, poiquilocitosis, células en blanco de tiro (codocitos). Hb A 2 3-8%. Hb F 1-5%. Talasemia mayor. Anemia Hb 3-4 g/dL. Hb F y Hb A 2 muy elevadas. Hemocromatosis. 1. Portador silencioso (casi normal). 2. Talasemia menor moderada anemia. Al nacer Hb Bart ( 4) 2-10% la que se pierde en el adulto. 3. Enfermedad HbH: Anemia Hb 7-10 g/dL. Los GR muestran inclusiones granulares finas (tinción supravital) como pelotas de golf. 4. Hidrops fetal ( 4): Incompatible con la vida.
. Heterocigotas (talasemia menor). Talasemia 4 variantes: Talasemia menor. Moderada anemia hemolítica y sideroblastica, hipocromia y microcitosis. Hb g/dL, poiquilocitosis, células en blanco de tiro (codocitos). Hb A 2 3-8%. Hb F 1-5%. Talasemia mayor. Anemia Hb 3-4 g/dL. Hb F y Hb A 2 muy elevadas. Hemocromatosis. 1. Portador silencioso (casi normal). 2. Talasemia menor moderada anemia. Al nacer Hb Bart ( 4) 2-10% la que se pierde en el adulto. 3. Enfermedad HbH: Anemia Hb 7-10 g/dL. Los GR muestran inclusiones granulares finas (tinción supravital) como pelotas de golf. 4. Hidrops fetal ( 4): Incompatible con la vida..")
70
D. DEFICIENCIAS ENZIMATICAS DEL GR. 1.DEFICIENCIA DE PIRUVATOCINASA PC ( RARA ). Prueba diagnóstica: Medición cuantitativa de la enzima. Tratamiento: Sintomático. Esplenectomía. TMO. 2. DEFICIENCIA DE GLUCOSA-6-FOSFATO DESHIDROGENASA G-6-PD. Defecto del GR: Es la mas frecuente del ciclo de las pentosas. Hay mas de 300 variantes bioquímicas. Defecto del GR: Es la más común del ciclo de Embden-Meyerhof. Herencia: Autosómica, dominante. Clínica: Anemia, ictericia, esplenomegalia. Hb 6-12 g/dL. Herencia: Ligada al sexo, recesiva. Raza: judía y negros Clínica: Anemia hemolítica congénita no esferocítica más frecuente. Individuo aparentemente sano ingiere reductores de G-6-PD y tiene crisis hemólitica intravascular. Favismo. Dx: GR con cuerpos de Heinz. Cuantificación de la enzima. Tratamiento: Sintomático. Esplenectomia. TMO.

71
DEFECTOS INTRACORPUSCULARES ADQUIRIDOS HEMOGLOBINURIA PAROXISTICA NOCTURNA HPN. Enfermedad clonal adquirida de célula madre hematopoyética. Gen PIG-A controla la glicosilphosphatidilinositol, proteína de anclaje en la superficie de la membrana para el inhibidor de membrana a la lisis reactiva (MIRL) CD59 y el factor acelerador del decaimiento (DAF) CD55, haciendo a los GR, GB y plaquetas sensibles al complemento principalmente en la noche. El paciente tiene 2 poblaciones de células: normales y HPN. Clasificación según el grado de hemólisis: HPN-I casi normal. HPN-II sensible a C 2-5 veces más (Leve). HPN-III 15-25 veces, cursa con anemia, hemosiderinuria, hemoglobinuria, deficiencias de Fe y Ac. fólico, trombocitopénia, infecciones y trombosis. Muta a AnemiaAplastica un 20% y rara vez a SMD o LAM. Pruebas diagnósticas:Ac Mo CD55 y CD59 bajos. HAM, sucrosa, inulina positivas. Acetilcolinesterasa baja. Sensibilidad al C+. Tratamiento: Sintomático. TMO.
 CD59 y el factor acelerador del decaimiento (DAF) CD55, haciendo a los GR, GB y plaquetas sensibles al complemento principalmente en la noche. El paciente tiene 2 poblaciones de células: normales y HPN. Clasificación según el grado de hemólisis: HPN-I casi normal. HPN-II sensible a C 2-5 veces más (Leve). HPN-III veces, cursa con anemia, hemosiderinuria, hemoglobinuria, deficiencias de Fe y Ac. fólico, trombocitopénia, infecciones y trombosis. Muta a AnemiaAplastica un 20% y rara vez a SMD o LAM. Pruebas diagnósticas:Ac Mo CD55 y CD59 bajos. HAM, sucrosa, inulina positivas. Acetilcolinesterasa baja. Sensibilidad al C+. Tratamiento: Sintomático. TMO..")
72
HEMOLISIS DE CAUSA EXTRACORPUSCULAR. A. VASCULAR (Microangiopática): Prótesis, estenosis, hemangiomas, circuitos AV, circulación extracorporea, microtrombos. GR: ezquistocitos, Cel. de Burr, poiquilocitos. B. HIPERESPLENISMO: Pancitopénia, esplenomegalia, MO NL. C. AUTOINMUNE PRIMARIA*: Ac vs GR. GR opsonizados. Prueba de Coombs positiva a 37ºC o menos. Tx: Corticoesteroides, esplenectomía, inmunosupresores. D. AUTOINMUNE SECUNDARIA*: Similar a la anterior pero de causada conocida: LES, LLC, medicamentos. E. ISOINMUNE*: Transfusiones. Anemia hemolítica del RN. *Casi todas la anemias hemolíticas inmunes cursan con P. de Coombs +.
: Prótesis, estenosis, hemangiomas, circuitos AV, circulación extracorporea, microtrombos. GR: ezquistocitos, Cel. de Burr, poiquilocitos. B. HIPERESPLENISMO: Pancitopénia, esplenomegalia, MO NL. C. AUTOINMUNE PRIMARIA*: Ac vs GR. GR opsonizados. Prueba de Coombs positiva a 37ºC o menos. Tx: Corticoesteroides, esplenectomía, inmunosupresores. D. AUTOINMUNE SECUNDARIA*: Similar a la anterior pero de causada conocida: LES, LLC, medicamentos. E. ISOINMUNE*: Transfusiones. Anemia hemolítica del RN. *Casi todas la anemias hemolíticas inmunes cursan con P. de Coombs +..")
74
ANEMIA DE LA ENFERMEDAD CRÓNICA

75
Características: Mala respuesta a los hematínicos y se corrige cuando la enfermedad de base se cura. Etiología: Multifactorial. 1.Liberación de IL1 (monocina) por células del SRE. Es mediadora de la respuesta de fase aguda. Produce rápida caída de Fe plasmático. Estimula la hemólisis por fagocitosis. 2.Liberación de lactoferrina por gránulos de los neutrófilos. Se liga al Fe y fagocitada por los macrófagos no lo libera. 3.Lactoferrina es bloqueadora de la hematopoyesis. 4.El Interferón es bloqueador de la hematopoyesis. 5.Factor de necrosis tumoral, bloquea hematopoyesis e induce mayor hemólisis.
 por células del SRE. Es mediadora de la respuesta de fase aguda. Produce rápida caída de Fe plasmático. Estimula la hemólisis por fagocitosis. 2.Liberación de lactoferrina por gránulos de los neutrófilos. Se liga al Fe y fagocitada por los macrófagos no lo libera. 3.Lactoferrina es bloqueadora de la hematopoyesis. 4.El Interferón es bloqueador de la hematopoyesis. 5.Factor de necrosis tumoral, bloquea hematopoyesis e induce mayor hemólisis..")
76
ANEMIA DE LA ENFERMEDAD CRÓNICA. DIAGNOSTICO: 1.El de la enfermedad crónica de base. 2.Cursa con anemia N.N. En > del 80 %, el resto puede ser hipocrómica microcítica o macrocítica. 3.Reticulocitos normales (0.5 – 2.0 %). 4.Protoporfirina libre aumentada (> 62 g/dL). 5.Ferritina sérica elevada (> 100 g/L). 6.Fe sérico bajo (< 50 g/dL) y capacidad total de fijación reducida (< 250 g/dL). 7. Tinción de Fe en MO: Fe extramedular aumentado, intracelular (eritroblastos) disminuido o ausente (anemia sideroblástica). Tratamiento: El de la enfermedad de base. Además Fe, Ac. Fólico, V. B12, Eritropoyetina, transfusiones de GR.
. 4.Protoporfirina libre aumentada (> 62 g/dL). 5.Ferritina sérica elevada (> 100 g/L). 6.Fe sérico bajo (< 50 g/dL) y capacidad total de fijación reducida (< 250 g/dL). 7. Tinción de Fe en MO: Fe extramedular aumentado, intracelular (eritroblastos) disminuido o ausente (anemia sideroblástica). Tratamiento: El de la enfermedad de base. Además Fe, Ac. Fólico, V. B12, Eritropoyetina, transfusiones de GR..")
Presentaciones similares















>")




. Forma primaria: herencia autosómica recesiva Alteración.>")

 y electrones (citocromos)>")


