Descargar la presentación
La descarga está en progreso. Por favor, espere

2
PROGRAMA DE BACTERIOLOGIA DIEGO FERNANDO LOPEZ MUÑOZ DOCENTE
FACULTAD DE SALUD PROGRAMA DE BACTERIOLOGIA DIEGO FERNANDO LOPEZ MUÑOZ DOCENTE HEMATOLOGÍA SÍNDROMES MIELOPROLIFERATIVOS

3
Síndromes Mieloproliferativos
El síndrome mieloproliferativo crónico es un conjunto heterogéneo de neoplasias hematológicas que tienen como característica común la proliferación descontrolada de los precursores medulares de alguna de las células sanguíneas animales, incluyendo los humanos

4
Por lo común, afectan la maduración de dichos precursores en fases avanzadas de la diferenciación, como dato diferencial con respecto a los síndromes mieloproliferativos agudos fases tempranas . El concepto de enfermedad mieloproliferativa fue propuesto por primera vez en 1951, por el hematólogo William Dameshek.

5
Trastornos mieloproliferativos (TMP) Síndromes mielodisplásicos (SMD)
Los trastornos neoplásicos de las células hematopoyéticas de la médula ósea (MO), se pueden agrupar generalmente en tres categorías principales, según la Organización Mundial de la Salud (OMS): Trastornos mieloproliferativos (TMP) Síndromes mielodisplásicos (SMD) Trastornos mieloproliferativos / síndromes mielodisplásicos como (leucemia mielomonocítica crónica) Los TMP y SMD, se caracterizan por la proliferación neoplásica autónoma de precursores hematopoyéticos. Los TMP generalmente pueden distinguirse de los SMD porque en aquellos, la sangre periférica muestra aumento en los eritrocitos, los leucocitos o las plaquetas.
, se pueden agrupar generalmente en tres categorías principales, según la Organización Mundial de la Salud (OMS): Trastornos mieloproliferativos (TMP) Síndromes mielodisplásicos (SMD) Trastornos mieloproliferativos / síndromes mielodisplásicos como (leucemia mielomonocítica crónica) Los TMP y SMD, se caracterizan por la proliferación neoplásica autónoma de precursores hematopoyéticos. Los TMP generalmente pueden distinguirse de los SMD porque en aquellos, la sangre periférica muestra aumento en los eritrocitos, los leucocitos o las plaquetas.")
6
Leucemia mieloide crónica. Leucemia granulocítica crónica.
Clasificación Leucemia mieloide crónica. Leucemia granulocítica crónica. Policitemia vera. Mielofibrosis con metaplasia mieloide. Trombocitemia esencial. La agrupación de estos procesos hematológicos se debe a que en todos la fisiopatología depende del trastorno en el crecimiento clonal expansivo de una célula progenitora hematopoyética denominada pluripontencial, cuyo resultado es la sobreproducción de uno o de varios de los elementos celulares circulantes en el torrente sanguíneo

7
Cromosoma Filadelfia "positiva" Cromosoma Filadelfia "negativa"
La leucemia mieloide crónica se caracteriza, a diferencia de los demás trastornos mieloproliferativos, por asociarse con un intercambio por translocación en el material genético entre el cromosoma 9 y el cromosoma 22 o t(9:22), lo cual produce el denominado cromosoma Filadelfia y expresa una proteína única denominada bcr-abl. Basado en ello, el síndrome mieloproliferativo crónico puede ser categorizado según la presencia o no del cromosoma Filadelfia Cromosoma Filadelfia "positiva" Cromosoma Filadelfia "negativa" Leucemia mieloide crónica (LMC) Policitemia vera(PV) Trombocitosis esencial (TE) Mielofibrosis (MF)
, lo cual produce el denominado cromosoma Filadelfia y expresa una proteína única denominada bcr-abl. Basado en ello, el síndrome mieloproliferativo crónico puede ser categorizado según la presencia o no del cromosoma Filadelfia. Cromosoma Filadelfia positiva Cromosoma Filadelfia negativa Leucemia mieloide crónica (LMC) Policitemia vera(PV) Trombocitosis esencial (TE) Mielofibrosis (MF)")
8
POLICITEMIAS RUBRA VERA Síndrome mieloproliferativo crónico (clonal)
POLI = MUCHOS CITO = CELULAS HEMO = SANGRE Síndrome mieloproliferativo crónico (clonal) Características: Eritrocitosis Aumento de masa globular Leucocitosis, trombocitosis, esplenomegalia Incidencia / habitantes Mayor frecuencia en hombres, 60 a 80 años El potencial proliferativo no parece afectar los progenitores de células T y NK. Es debida a un exceso de sensibilidad a la eritropoyetina de los precursores de la medula ósea.
 Características: Eritrocitosis. Aumento de masa globular. Leucocitosis, trombocitosis, esplenomegalia. Incidencia / habitantes. Mayor frecuencia en hombres, 60 a 80 años. El potencial proliferativo no parece afectar los progenitores de células T y NK. Es debida a un exceso de sensibilidad a la eritropoyetina de los precursores de la medula ósea.")
9
ESP paciente con PRV Mieloproliferación (aumento de serie roja)
Hiperviscosidad sanguínea, hipermetabolismo, hemorragias y trombosis Esplenomegalia y hepatomegalia Aumento del numero de hematíes (VCM disminuido) Leucocitosis (neutrófilos: aumento de fosfatasa alcalina)
 Leucocitosis (neutrófilos: aumento de fosfatasa alcalina)")
10
Cuadro clínico Policitemia
Mieloproliferación (aumento de serie roja) Disminución de eritropoyetina Aumento de vitamina B12 Trombocitosis AMO: hiperplasia de las tres series (roja) Fibrosis de la médula Desaceleración en la producción de células (se puede llegar a producir anemia) y tendencia a fibrosis medular Se le conoce también como PV gastada
 Disminución de eritropoyetina. Aumento de vitamina B12. Trombocitosis. AMO: hiperplasia de las tres series (roja) Fibrosis de la médula. Desaceleración en la producción de células (se puede llegar a producir anemia) y tendencia a fibrosis medular. Se le conoce también como PV gastada.")
11
POLICITEMIA RUBRA VERA
Fibrosis de la médula Vida media sin tratamiento es de 2 años y con tratamiento unos 10 años Es una enfermedad incurable La causa de muerte mas frecuente es la trombosis Signos y síntomas mas frecuentes Por aumento de la masa globular y enlentecimeinto del flujo por hiperviscosidad: Cefalea, mareos, tinnitus, alteraciones visuales, disnea, debilidad, baja de peso, dolores óseos y articulares, plenitud abdominal, prurito. Rubicundez, lesiones cutáneas vasculares, urticariales, equimosis, cianosis, eritromegalia, esplenomegalia, inyección conjuntival, hepatomegalia, hipertensión arterial

12
Policitemia Vera: laboratorio
Hemoglobina 16 a 18 gr/dl Eritrocitos 7 a 10 x 1012 Leucocitosis con desviación a izquierda Aumento de eosinófilos y basófilos Trombocitosis Aumento de masa globular (51Cr) Se realiza con técnicas de dilución mediante el radioisótopo (Cr51) que marca una muestra de eritrocitos del paciente “in vitro”. Una masa > de 36 mg/Kg en hombres y > de 32mg/Kg en la mujer orientan a una policitemia absoluta. Eritropoyetina normal Viscosidad sanguínea aumentada Hiperuricemia Médula ósea hipercelular, sin grasa; hiperplasia de megacariocitos; depósitos de hierro bajos; aumento de fibras
 Se realiza con técnicas de dilución mediante el radioisótopo (Cr51) que marca una muestra de eritrocitos del paciente in vitro . Una masa > de 36 mg/Kg en hombres y > de 32mg/Kg en la mujer orientan a una policitemia absoluta. Eritropoyetina normal. Viscosidad sanguínea aumentada. Hiperuricemia. Médula ósea hipercelular, sin grasa; hiperplasia de megacariocitos; depósitos de hierro bajos; aumento de fibras.")
14
Policitemia Vera: criterios diagnósticos
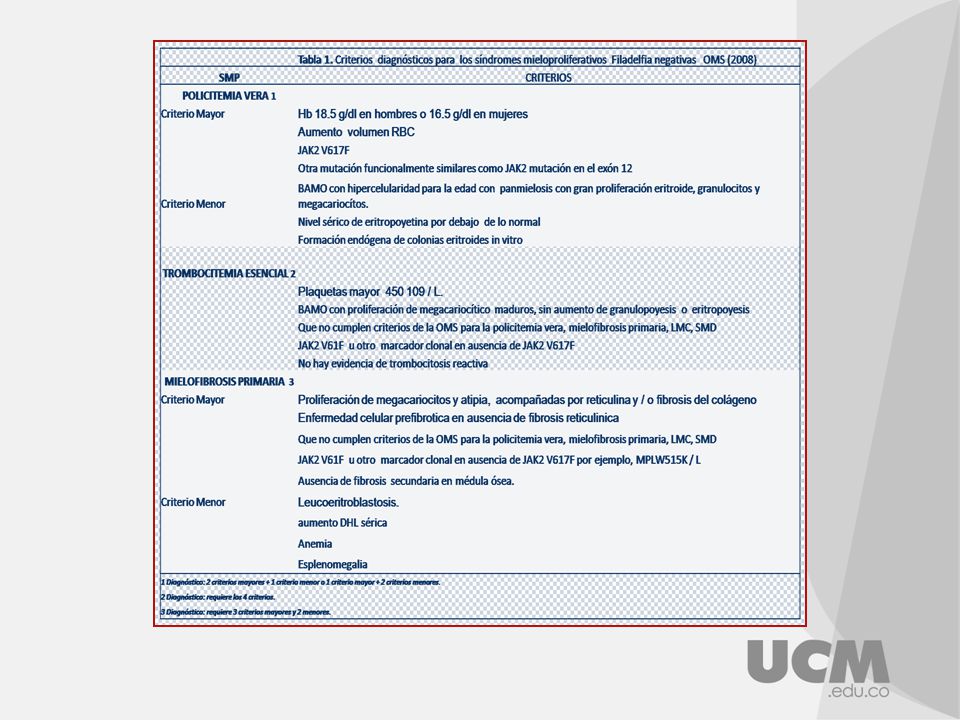
Masa globular ♂> 36 ml/kg ♀> 32 ml/kg saturation arterial de O2 >92% Esplenomegalia Trombocitosis > /l Leucocitosis > /l Fosfatasa alcalina leucocitaria > 100 B12 sérica > 900 pg/ml capacidad de unión a B12 insaturada >2200 pg/ml JAK (V617F), que parece sensibilizar a los precursores eritroides a la acción de la eritropoyetina. La policitemia vera es de causa desconocida aunque se ha observado en ciertos estudios que en el 95% de los pacientes afectados por la enfermedad, sufren una mutación en el gen de la tirosín-kinasa
, que parece sensibilizar a los precursores eritroides a la acción de la eritropoyetina. La policitemia vera es de causa desconocida aunque se ha observado en ciertos estudios que en el 95% de los pacientes afectados por la enfermedad, sufren una mutación en el gen de la tirosín-kinasa.")
15
POLICITEMIA VERA: ETAPAS DE LA ENFERMEDAD
Asintomático : Esplenomegalia Eritrocitosis / trombocitosis aislada Fase eritrocítica : Eritrocitosis Trombocitosis Leucocitosis Esplenomegalia Trombosis / hemorragia / Prurito Fase inactiva : Masa eritrocitaria sin variación Metaplasia mieloide: 10% a los 10 años, 50% a los 20 años Anemia, Mayor esplenomegalia Reacción leucoeritroblástica Leucemia aguda : Generalmente mieloide

16
Policitemia Vera: evolución
Causas de muerte: Trombosis y tromboembolismo Leucemia aguda Otra neoplasia Hemorragia Mielofibrosis

17
Tratamiento Y Pronostico
Hay diferentes opciones de tratamiento que implican tipos de complicaciones particulares; sin embargo, la única potencialmente curativa es el trasplante de células hematopoyéticas. Flebotomías Quimioterapia con hidroxiurea (busulfan, clorambucil, melfalán) Terapéutica antitrombótica
 Terapéutica antitrombótica.")
18
¿Cuál es la función normal del gen JAK2?
Proporciona instrucciones para hacer una proteína que promueve el crecimiento y la división (proliferación) de las células. Esta proteína es parte de una vía de señalización de llamada de la vía JAK / STAT, que transmite señales químicas de fuera de la célula al núcleo de la célula. La proteína JAK2 es especialmente importante para el control de la producción de células sanguíneas a partir de células madre hematopoyéticas. Estas células madre se encuentran dentro de la médula ósea y tienen el potencial de convertirse en glóbulos rojos, glóbulos blancos y plaquetas Janus kinase 2 (comúnmente llamada JAK2) es una proteína que está implicada en la señalización de: Interferón, UFC-GM, IL-3R, IL-5R, IL-6R, EPO-R,TPO-R, R-UFC-GM. Proliferación aumentada, hipersensibilidad a las citoquinas, diferenciación independiente de citoquinas e inhibición de la apoptosis.
 de las células. Esta proteína es parte de una vía de señalización de llamada de la vía JAK / STAT, que transmite señales químicas de fuera de la célula al núcleo de la célula. La proteína JAK2 es especialmente importante para el control de la producción de células sanguíneas a partir de células madre hematopoyéticas. Estas células madre se encuentran dentro de la médula ósea y tienen el potencial de convertirse en glóbulos rojos, glóbulos blancos y plaquetas. Janus kinase 2 (comúnmente llamada JAK2) es una proteína que está implicada en la señalización de: Interferón, UFC-GM, IL-3R, IL-5R, IL-6R, EPO-R,TPO-R, R-UFC-GM. Proliferación aumentada, hipersensibilidad a las citoquinas, diferenciación independiente de citoquinas e inhibición de la apoptosis.")
19
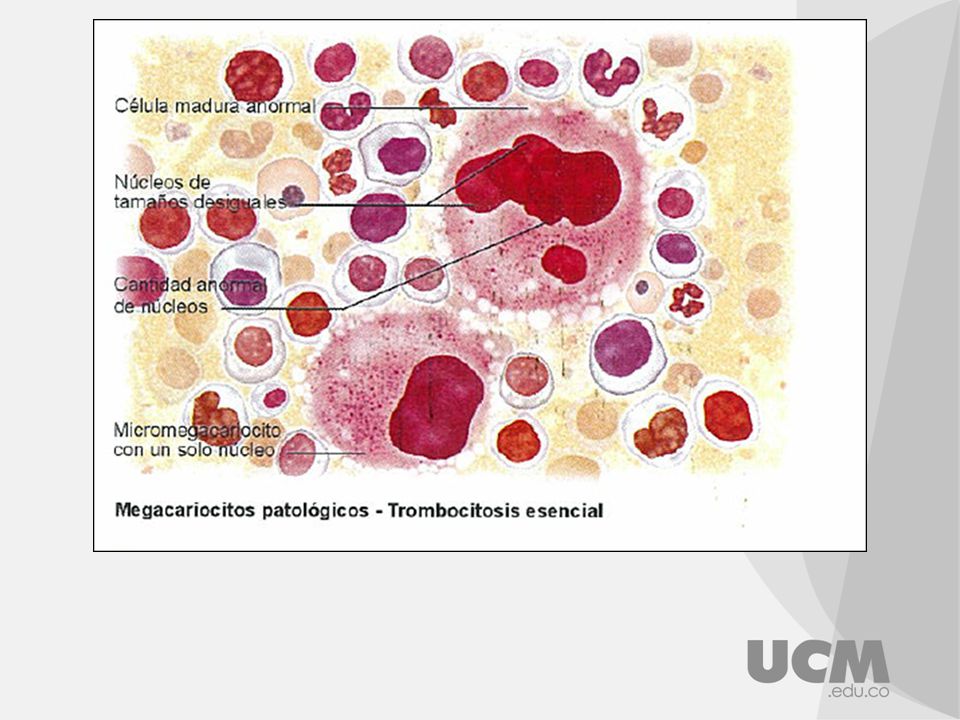
TROMBOCITEMIA ESENCIAL
Pertenece a los SMP Proliferación de megacariocitos y exceso de plaquetas circulantes Descrita en 1934, por Epstein y Goedel Considerada desde 1951, como SMP Trastorno mieloproliferativo de origen clonal de las células pluripotenciales, tiene expresión fenotípica, sobre toda la línea megacariocitica y plaquetas, pero afecta a todas las células sanguíneas.

20
Etiopatogenia: Se desconoce la causa de la trombocitosis esencial, si bien se ha demostrado origen clonal. Esta enfermedad suele presentarse en personas entre los 50 y 70 años y afecta en igual proporción a mujeres y hombres. Características: Las plaquetas sufren diversas anomalías morfológicas Bioquímicas Funcionales: ya que se observan disminución o ausencia de los gránulos citoplasmáticos junto con alteraciones del tamaño y volumen plaquetario. En la agregometria plaquetaria se advierte una atenuación o falta de respuesta a los agonistas plaquetarios usuales, como ADP, Trombina y colágeno.

21
Cuadro clínico: Manifestaciones hemorrágicas del tubo digestivo y la mucosa nasal. Tromboticas: Trombosis arterial lo cual conduce a ataques isquémicos transitorios o incluso a infarto cerebral, isquemia coronaria. Hallazgo mas común es la esplenomegalia, además de equimosis y eventos hemorrágicos. Diagnostico: recuento plaquetario alto en la mayoría de los pacientes > /Ul. En el frotis de sangre periférica se observa anisotrombia y agregados plaquetarios. Aspirado de MO: hiperplasia megacariocitica. La mutación V617F del gen JAK-2 hasta en 50% de los casos y si aparece positivo es un indicador de mal pronostico

22
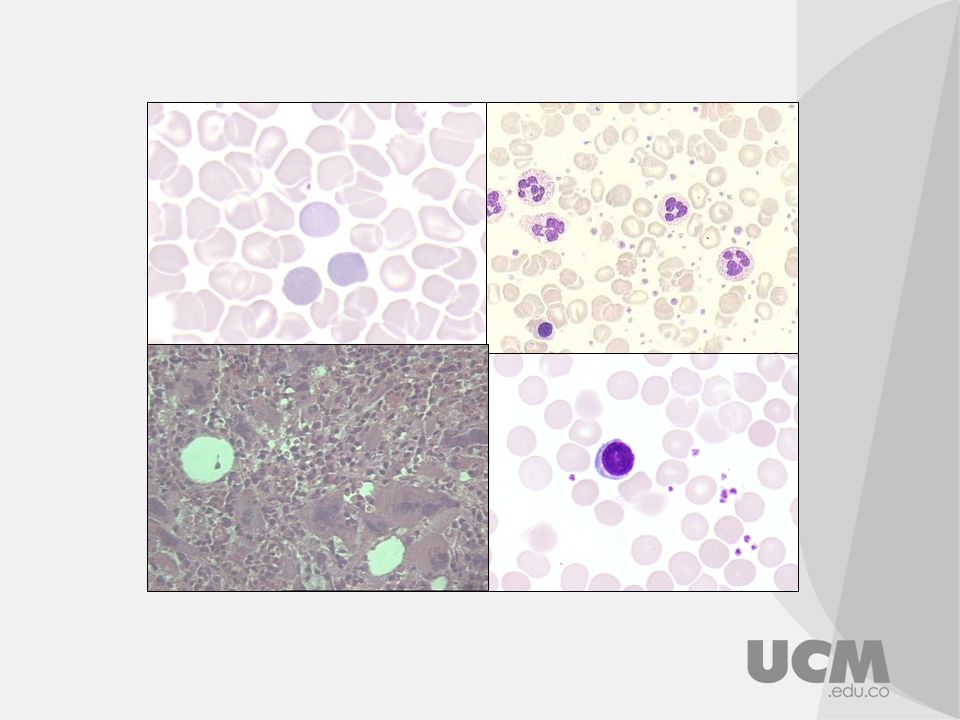
Aspirado de médula ósea de un paciente con ET y JAK2 V167F positivo, se observan megacariocitos grandes e hiperlobulados (Teñido con Wright-Giemsa)
")
23
Manifestaciones clínicas
Trombocitemia primaria Gangrena acrocianótica periférica AIT atípico Enfermedad coronaria Trombo plaquetario transitorio u oclusivo en arteriolas Plaquetas activadas Secreción, agregación Endoperóxidos de prostaglandinas, pero no tromboxano A2 Inflamación no específica y congestión Factor de crecimiento derivado de las plaquetas Proliferación de músculo liso. Proliferación de la capa fibromuscular íntima de arteriolas AIT= Ataque isquémico transitorio

26
Manifestaciones clínicas
Frecuencia de manifestaciones neurológicas asociadas con trombocitemia esencial Manifestaciones Pacientes (N) Cefalea 13 Parestesias 10 Isquemia cerebral posterior 9 Isquemia cerebral anterior 6 Perturbaciones visuales Convulsiones 2 No. total de pacientes 33
 Cefalea. 13. Parestesias. 10. Isquemia cerebral posterior. 9. Isquemia cerebral anterior. 6. Perturbaciones visuales. Convulsiones. 2. No. total de pacientes. 33.")
27
Hallazgos de laboratorio asociados con trombocitemia esencial
No. de pacientes % Nivel de hemoglobina >16 g/100 mL 6 12-16 g/dL 64 68 <12 g/dL 23 24 Leucocitosis <8 x 109/L 12-20 x 109/L 41 44 20-29 x 109/L 22 Cuenta plaquetaria <1 x 1012/L 37 38 x 1012/L 39 >1.5 x 1012/L 20 Fibrosis reticularia 38/70 54 Citogenénica normal 49/51 99 Agregación plaquetaria Normal 4/64 Disminuido Después de APA 19/64 30 Después de colágeno 6/64 9 Después de ADP y colágeno 35/64 55

28
Criterio para el diagnóstico de la trombocitemia esencial
1. Cuenta plaquetaria >600,000/mm3 en 2 ocasiones diferentes con 1 mes de intervalo 2. Ausencia de causa identificable de trombosis reactiva 3. Volumen eritrocitario normal 4. Ausencia de fibrosis significativa de la médula 5. Ausencia del cromosoma Ph y de la fusión del gen BCR/ABL por PCR; ausencia de anormalidades citogenéticas clonales asociadas con trastornos mielodisplásicos 6. Presencia de esplenomegalia a la exploración física, ultrasonido y CT scan 7. Hipercelularidad de la médula ósea en la biopsia y presencia de hiperplasia megacariocítica con líneas de grandes megacariocitos multilobulares 8. Ausencia de deficiencia de hierro documentada por función de hierro medular y/o fenitina sérica normal 9. En mujeres, demostración de hematopogesis clonal, por medio de análisis de longitud de fragmentos de restricción de genes presentes en el cromosoma x 10. Presencia de células progenitoras anormales en médula determinada por la formación de eritroide endógeno y/o colonias megacariocíticas con sensibilidad incrementada a IL-3 11. Ausencia de elevación proteína C=reactiva en plasma y de niveles de IL-6. Pacientes que cumplan los criterios del 1 al 5 y ≥3 de los criterios del 6 al 11 deben considerarse con diagnóstico de trombocitemia primaria

29
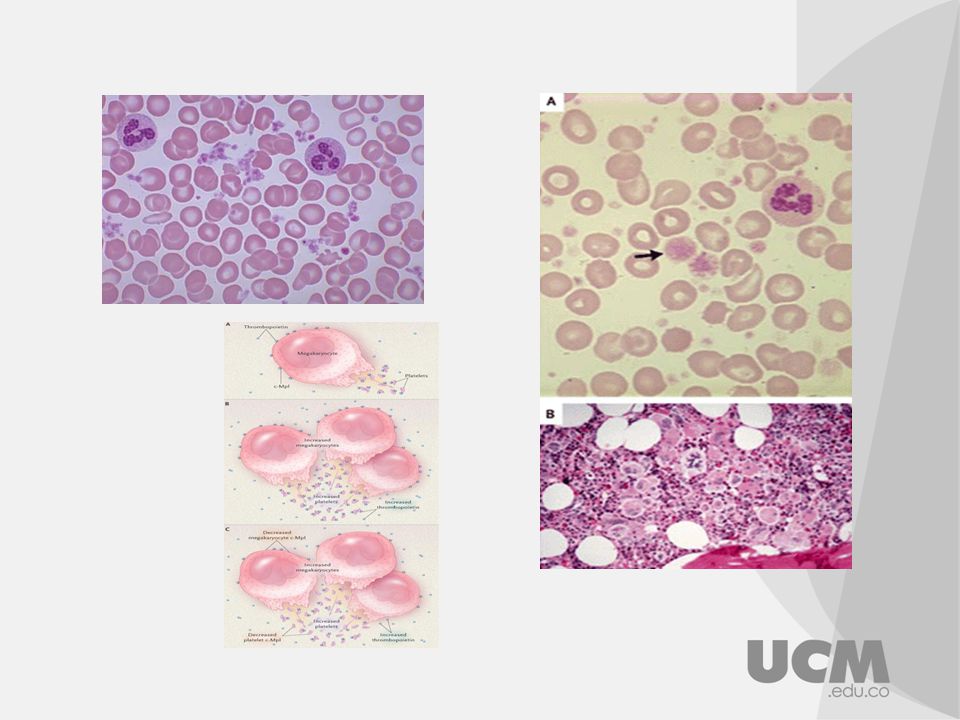
Mielofibrosis: Conocida como metaplasia mieloide agnogena, neoplasia mieloproliferativa que tiene su origen en las células precursoras hematopoyéticas, se caracteriza por la presencia de granulocitos inmaduros, precursores eritroides, dacriocitos, con grados variables de fibrosis en MO y esplenomegalia. El nombre se refiere al deposito excesivo de colágeno en MO. Cuadro clínico: Anemia que se manifiesta con astenia, palidez, palpitaciones disnea de esfuerzos. Estado hipermetabolico con febrícula, diaforesis y perdida de peso. Esplenomegalia con dolor en hipocondrio izquierdo (90% pacientes)
")
30
Hallazgos de laboratorio
Anemia normocitica normocromica manifestación mas frecuente 80% casos También es característico reacción leucoeritroblastica es decir la presencia de eritroblastos y células mieloides inmaduras como mielocitos y metamielocitos, puede encontrarse trombocitopenia o trombocitosis. Perfil bioquímico aumento de la LDH. Aspirado de MO es difícil y en muchas ocasiones es una punción seca En el inicio de la enfermedad en MO se observa: hiperplasia eritroide, granulocitica y megacariocitica. Diagnostico: La mutación V617F del gen JAK-2 hasta en un 50% de los casos y es una determinación útil en la confirmación del diagnostico y sobre todo de la elección de algunas opciones terapéuticas actuales.

31
Características clínicas: síntomas
Fatiga Debilidad Disnea Palpitaciones Pérdida de peso Saciedad temprana Dolor abdominal (cuadrante superior izq.) con irradiación a hombro izq. Hemorragia inesperada Dolor oseo (extremidad inferior) ¼ de pacientes son asintomáticosHepatomegalia 2/3 Esplenomegalia (casi todos)
 con irradiación a hombro izq. Hemorragia inesperada. Dolor oseo (extremidad inferior) ¼ de pacientes son asintomáticosHepatomegalia 2/3. Esplenomegalia (casi todos)")
32
Alessandro Vannucchi – Mario Cazzola
Rassegna stampa Alessandro Vannucchi – Mario Cazzola New England Journal of Medicine 10 dicembre 2013 Ansa Identificata nuova mutazione genica in tumori del sangue, studio UniFi sul New England Journal Il gruppo diretto da Alessandro Maria Vannucchi, del Dipartimento di Medicina Sperimentale e Clinica dell’Università di Firenze e Dipartimento di Oncologia dell’Azienda Ospedaliera Universitaria Careggi, ha contribuito ad una scoperta molto importante per i pazienti affetti da malattie mieloproliferative. Insieme a ricercatori dell’Università di Cambridge Vannucchi e i suoi collaboratori hanno identificato una mutazione nel gene della calreticulina (CALR) nella maggior parte dei pazienti affetti da due neoplasie mieloproliferative, la trombocitemia e la mielofibrosi primaria, che risultavano negative alla già nota e più comune mutazione del gene JAK2.
 nella maggior parte dei pazienti affetti da due neoplasie mieloproliferative, la trombocitemia e la mielofibrosi primaria, che risultavano negative alla già nota e più comune mutazione del gene JAK2.")
33
CALR – Trombocitemia Esencial, Mielofibrosis Primaria
Mutaciones en el gen CALR (calreticulina) Recientemente se ha demostrado que el gen que codifica la calreticulina (CALR) se encuentra mutado en la mayoría de los pacientes con un síndrome mieloproliferativo que carecen de una mutación en el gen JAK2. Se han encontrado una variedad de inserciones/delecciónes en el exón 9 del gen de la calreticulina en la mayoría (~70-85%) de las trombocitemias esenciales y en mielofibrosis primarias que no tengan JAK2 y MPL (receptor natural de plaquetas) mutados, y en una minoría de pacientes con mielodisplasia (8%).
 Recientemente se ha demostrado que el gen que codifica la calreticulina (CALR) se encuentra mutado en la mayoría de los pacientes con un síndrome mieloproliferativo que carecen de una mutación en el gen JAK2. Se han encontrado una variedad de inserciones/delecciónes en el exón 9 del gen de la calreticulina en la mayoría (~70-85%) de las trombocitemias esenciales y en mielofibrosis primarias que no tengan JAK2 y MPL (receptor natural de plaquetas) mutados, y en una minoría de pacientes con mielodisplasia (8%).")
34
La disponibilidad de este nuevo análisis de diagnóstico clínico para estas mutaciones comunes en el exón 9 del gen de la calreticulina, junto con el análisis del gen JAK2 para la mutación V617F (y en menor grado mutaciones en MPL), permite identificar y controlar un nuevo marcador molecular específico en la gran mayoría (>90%) de los pacientes con una neoplasia mieloproliferativa . la expresión in vitro de esta mutación transmite crecimiento de citoquinas independiente, de señalización.

35
CALR en Policitemia Vera
No han sido reportadas mutaciones en CALR en pacientes con PV, razón por la cual el estudio molecular puede ser utilizado para distinguir pacientes con PV de aquellos que presentan TE o MP. JAK2 POSITIVO CARLP NEGATIVO JAK2 NEGATIVO CARLP POSITIVO EXÓN 9

36
MEDULA ÓSEA CON MIELOFIBROSIS

39
Leucemia Mieloide Crónica
Proliferación incontrolada de células mieloides. Se inicia en células madre hematopoyéticas pluripotenciales, que dan lugar a la proliferación preferente de los progenitores de la serie granulocítica.

40
CARACTERÍSTICAS DE LA LMC
15-20% de todas las leucemias en adultos Incidencia de 1.3/100,000/año Relación hombre/mujer 1.7:1 Mediana de edad 50 años (5% > 70 años; raro en la infancia) No existe correlación etiológica con exposición a mielotóxicos y no tiene etiología viral
 No existe correlación etiológica con exposición a mielotóxicos y no tiene etiología viral.")
41
Fisiopatología Esta presente en los granulocitos, eritrocitos y precursores plaquetarios, en > 95% de los pacientes con LMC. El defecto genético del cromosoma Philadelfia consiste en un fenómeno conocido como TRASLOCACIÓN, en donde partes de dos cromosomas, el 9 y el 22(traslocacion 9-22), intercambian sus posiciones. El resultado es que parte del gen de región de fractura (BCR, Breakpoint Cluster Región) del cromosoma 22 (región q11) se fusiona con parte del gen ABL del cromosoma 9 (región q34). La proteína resultante de la fusión BCR-ABL interactúa con la subunidad receptora interleuquina 3beta(c). La transcripción del BCR-ABL permanece activa continuamente, sin necesidad de ser activado por otras proteínas mensajeras. Éste, a su vez, activa un número de proteínas y enzimas controladoras del ciclo de división celular e inhibe la reparación del ADN, lo cual ocasiona la inestabilidad del genoma.
, intercambian sus posiciones. El resultado es que parte del gen de región de fractura (BCR, Breakpoint Cluster Región) del cromosoma 22 (región q11) se fusiona con parte del gen ABL del cromosoma 9 (región q34). La proteína resultante de la fusión BCR-ABL interactúa con la subunidad receptora interleuquina 3beta(c). La transcripción del BCR-ABL permanece activa continuamente, sin necesidad de ser activado por otras proteínas mensajeras. Éste, a su vez, activa un número de proteínas y enzimas controladoras del ciclo de división celular e inhibe la reparación del ADN, lo cual ocasiona la inestabilidad del genoma.")
42
La translocación da lugar a la fusión de una parte del gen bcr situado en el cromosoma 22, con el gen abl del cromosoma 9. El gen de fusión bcr-abl dirige la síntesis de proteína de 210 Kd que posee actividad de tirosina quinasa. Funciones: La transducción de señales Las hormonas que actúan sobre los receptores asociados a tirosina quinasas son generalmente factores de crecimiento que promueven la división celular.

43
Molestias abdominales
Cuadro clínico Síntomas Frecuencia Origen Constitucionales Febrícula, sudoración, pérdida de peso, anorexia 20% El hipermetabolismo secundario a la mieloproliferación Molestias abdominales Dolor en hipocondrio izquierdo, plenitud postprandial Esplenomegalia Síndrome anémico 10% Disminución en la producción de los glóbulos rojos por hiperplasia granulocítica Otros Dolor óseo, gota, hemorragia, priapismo <5% Infiltración medular, hiperuricemia, disfunción plaquetaria, hiperleucocitosis

44
Exploración física Esplenomegalia 50% Hepatomegalia 10-15% Palidez cutánea (según el grado de anemia) Otros: adenopatías, infiltración SNC <5

45
Exámenes de laboratorio
Leucocitosis granulocítica >100x109/L 50% <50 x 109/L 30% Basofilia Degranulación moderada de neutrófilos y pseudo-Pelger-Hüet Trombocitosis 45% Anemia habitualmente normocítica normocrómica Eritroblastos circulantes 30% Incremento en vitamina B12 100% Aumento LDH 80% Aumento ácido úrico 50%

46
Frotis en sangre Frotis en médula ósea
Cuadro clínico Leucemia mieloide crónica en fase crónica Frotis en sangre Frotis en médula ósea En el Aspirado de Médula Ósea se observa aumento en la relación mieloide/eritroide, hiperplasia granulocítica en todos los estadios de maduración, blastos habitualmente < 5%, hiperplasia megacariocítica. En la biopsia de hueso se observa aumento en la celularidad con disminución o ausencia de grasa, hiperplasia granulocítica y frecuentemente megacariocitos aumentados. Fibrosis en grado variable.

47
Diagnóstico diferencial
Comentarios Reacciones leucemoides Secundarias a infecciones o neoplasias Leucocitosis neutrofílica sin basofilia Cromosoma PH negativo Leucemia neutrofílica crónica Gammapatía monoclonal en 1/3 de los casos Síndrome hipereosinofílico Leucocitosis eosinofílica no explicable por otra causa, anemia y plaquetopenia frecuentes Posible reordenamiento FIP1L1/PDGFRA Formas proliferativas de LMMC Monocitosis en SP con promonocitos y rasgos displásicos en MO

48
Fases de LMC La LMC se diagnóstica en alrededor de 80% de los casos durante la fase crónica. 40% de estos pacientes se encuentra asintomático al momento de su diagnóstico. La fase crónica de la enfermedad tiene una duración aproximada 35 a 65 meses y dos tercios de los pacientes evolucionan a una fase denominada acelerada, que generalmente tiene una duración entre 12 y 24 meses. La última etapa corresponde a la fase blástica con una supervivencia de 3 a 12 meses. Se observa: Mieloide 60% Linfoide 25% Megacarioblástico 10-15% Eritroide 1%

49
Confirmación diagnóstica
Confirma la LMC: Presencia de cromosoma Filadelfia. Actividad reducida de fosfatasa alcalina leucocitaria. Dx Diferencial con: Leucemias agudas. Otras enfermedades Mieloproliferativas: mielofibrosis primaria, trombocitosis esencial, policitemia rubra vera. Leucemia monomielocítica crónica.

50
Fase Crónica: < 15% de blastos en SP y MO.
FASES DE LA ENFERMEDAD Fase Crónica: < 15% de blastos en SP y MO. Fase Acelerada :>15 %de blastos en SP y MO, pero <30% de blastos en ambos (SP y MO), >30% blastos mas Promielocitos en SP y MO. >20 % basófilos en SP. Trombocitopenia (< /mm3)no relacionado a la terapia. Fase Blástica:> de 30% de blastos en MO o SP, o localización extramedular (cloromas), excepto bazo e hígado.
, >30% blastos mas Promielocitos en SP y MO. >20 % basófilos en SP. Trombocitopenia (< /mm3)no relacionado a la terapia. Fase Blástica:> de 30% de blastos en MO o SP, o localización extramedular (cloromas), excepto bazo e hígado.")
51
Aceleración de la LMC a fase blástica
Eventos clínicos: Aparición de esplenomegalia (con infartos esplénicos). Aumento de la basofilia y la eosinofilia. Fiebre. Fibrosis medular. Resistencia a la hidroxiurea. * Fase de aceleración: ↑ % blastos y promielocitos.
. Aumento de la basofilia y la eosinofilia. Fiebre. Fibrosis medular. Resistencia a la hidroxiurea. * Fase de aceleración: ↑ % blastos y promielocitos.")
Presentaciones similares