Descargar la presentación
La descarga está en progreso. Por favor, espere

1
TRANSTORNOS DE LA CASCADA DE LA COAGULACION
FISIOPATOLOGIA PARA ENFERMERIA UNIDAD VII: Alteraciones Hematológicas

5
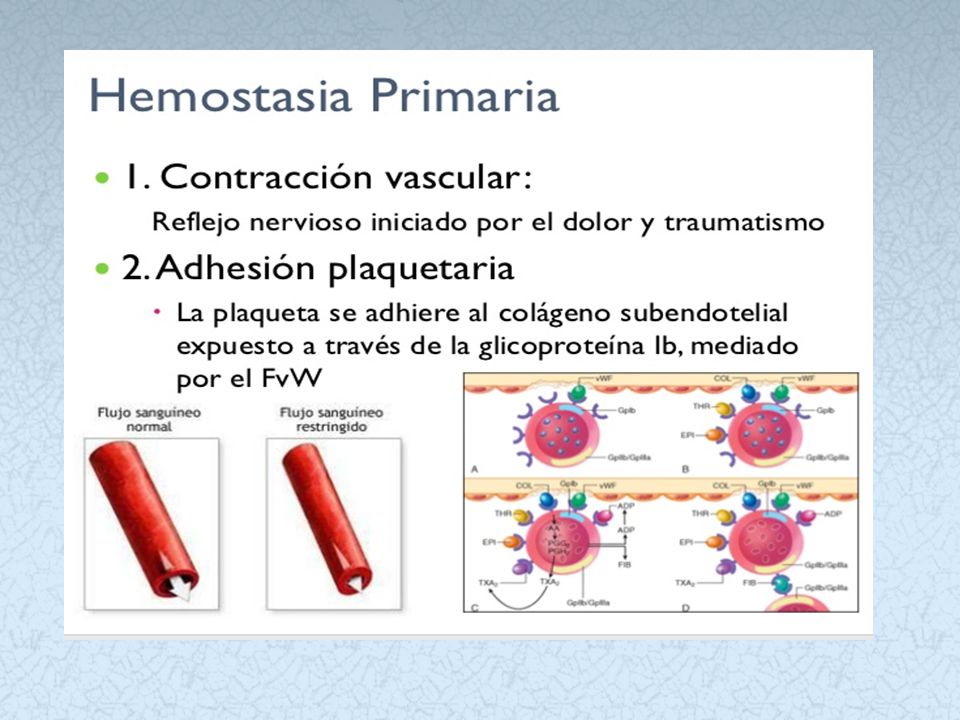
FUNCION DE LAS PLAQUETAS
Las plaquetas intervienen en el proceso de hemostasia. A nivel de la hemostasia primaria, trombo plaquetario. También en la hemostasia secundaria, a través de la liberación de factores.

6
FACTORES DE COAGULACION
Los factores de coagulación son un grupo de proteínas responsables de activar el proceso de coagulación. Hay identificados 13 factores ( I, II,...XIII). Los factores de coagulación actúan en cascada, es decir, uno activa al siguiente; si se es deficitario de un factor, no se produce la coagulación o se retrasa mucho.
. Los factores de coagulación actúan en cascada, es decir, uno activa al siguiente; si se es deficitario de un factor, no se produce la coagulación o se retrasa mucho.")
7
SINTESIS DE LOS FACTORES
Las plaquetas también sintetizan o contienen sustancias que intervienen en la coagulación del plasma sanguíneo. Factor plaquetario 3 (f3p): atrae a otros factores de la coagulación y facilita su reunión. Es necesario para iniciar la coagulación por la vía intrínseca Fibrinógeno: es la factor plasmático I de la coagulación. Fundamentalmente se encuentra en el plasma pero una pequeña parte de él está contenida en los gránulos alfa de las plaquetas y en la superficie de las mismas Factor Von Willebrand: una parte se encuentra en los gránulos alfa de las plaquetas. Contribuye a la adhesión y ayuda al mantenimiento plasmático del factor VIII-C de la coagulacón Factor V: Un 25% de la cantidad total se almacena en los gránulos plaquetarios. Factor plasmático XIII: se encuentra un porcentaje en las plaquetas que oscila entre el 30 y el 50%
: atrae a otros factores de la coagulación y facilita su reunión. Es necesario para iniciar la coagulación por la vía intrínseca. Fibrinógeno: es la factor plasmático I de la coagulación. Fundamentalmente se encuentra en el plasma pero una pequeña parte de él está contenida en los gránulos alfa de las plaquetas y en la superficie de las mismas. Factor Von Willebrand: una parte se encuentra en los gránulos alfa de las plaquetas. Contribuye a la adhesión y ayuda al mantenimiento plasmático del factor VIII-C de la coagulacón. Factor V: Un 25% de la cantidad total se almacena en los gránulos plaquetarios. Factor plasmático XIII: se encuentra un porcentaje en las plaquetas que oscila entre el 30 y el 50%")
12
FASES DE LA COAGULACION

19
ENFERMEDADES POR ALTERACION
DE LA COAGULACION

31
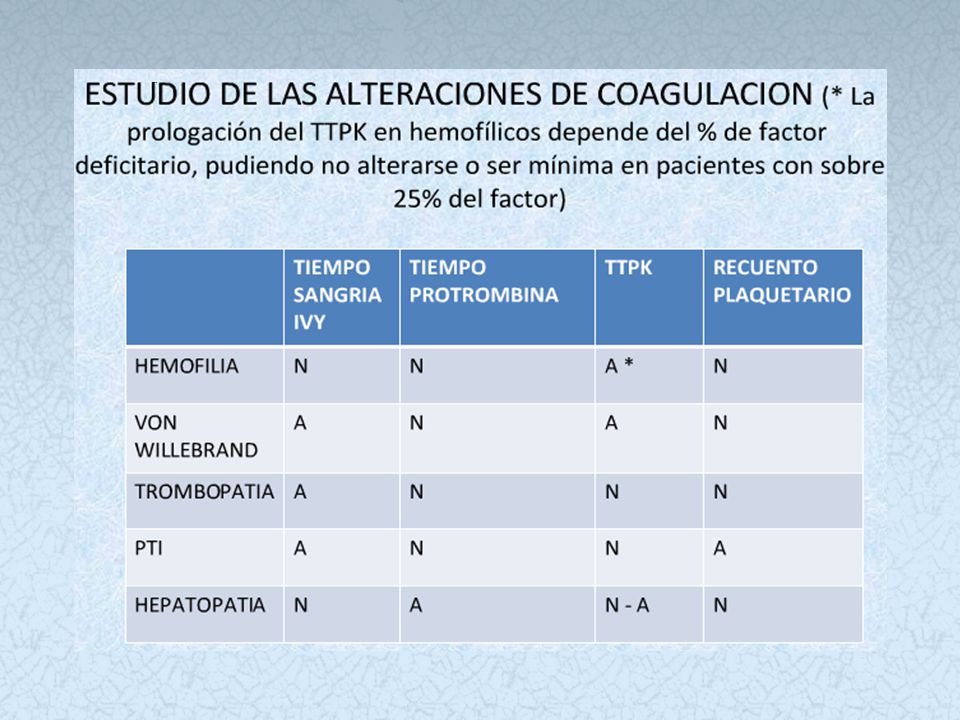
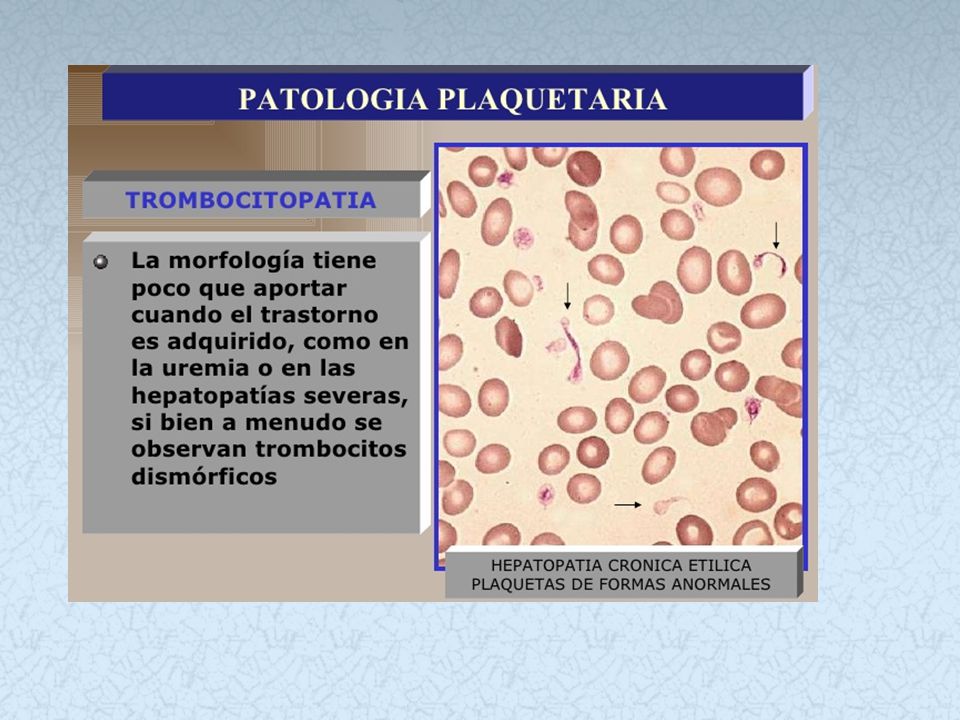
Diátesis Hemorrágica Trombopática
Normal: 275,000 plaquetas/μL (150, ,000 plaquetas/μL) TROMBOCITOSIS Hasta plaquetas/μL Ferropenia => estimulación hematopoyética elevada Esplenectomía: Falta de eliminación de plaquetas TROMBOCITEMIA Suele superar el plaquetas/μL Síndrome mieloproliferativo de los megacariocitos Plaquetas elevadas pero de aspecto y función anormales TROMBOPATIAS Plaquetas normales en cantidad pero funcionalmente anormales Alteración de la membrana Síndrome de Bernard-Soulier (falta gp-Ib=> falla adhesión) S. Glanzman o Tromboastenia (falta gp-IIb y gp-IIIa => falla agregación Alteración del contenido intracelular Síntesis de tromboxano A2 alterada (falla agregación y espasmo vascular) Disfunción asociada a enfermedad primaria Insuficiencia hepática, insuficiencia renal, fármacos, etc .
 TROMBOCITOSIS. Hasta plaquetas/μL. Ferropenia => estimulación hematopoyética elevada. Esplenectomía: Falta de eliminación de plaquetas. TROMBOCITEMIA. Suele superar el plaquetas/μL. Síndrome mieloproliferativo de los megacariocitos. Plaquetas elevadas pero de aspecto y función anormales. TROMBOPATIAS. Plaquetas normales en cantidad pero funcionalmente anormales. Alteración de la membrana. Síndrome de Bernard-Soulier (falta gp-Ib=> falla adhesión) S. Glanzman o Tromboastenia (falta gp-IIb y gp-IIIa => falla agregación Alteración del contenido intracelular. Síntesis de tromboxano A2 alterada (falla agregación y espasmo vascular) Disfunción asociada a enfermedad primaria. Insuficiencia hepática, insuficiencia renal, fármacos, etc .")
33
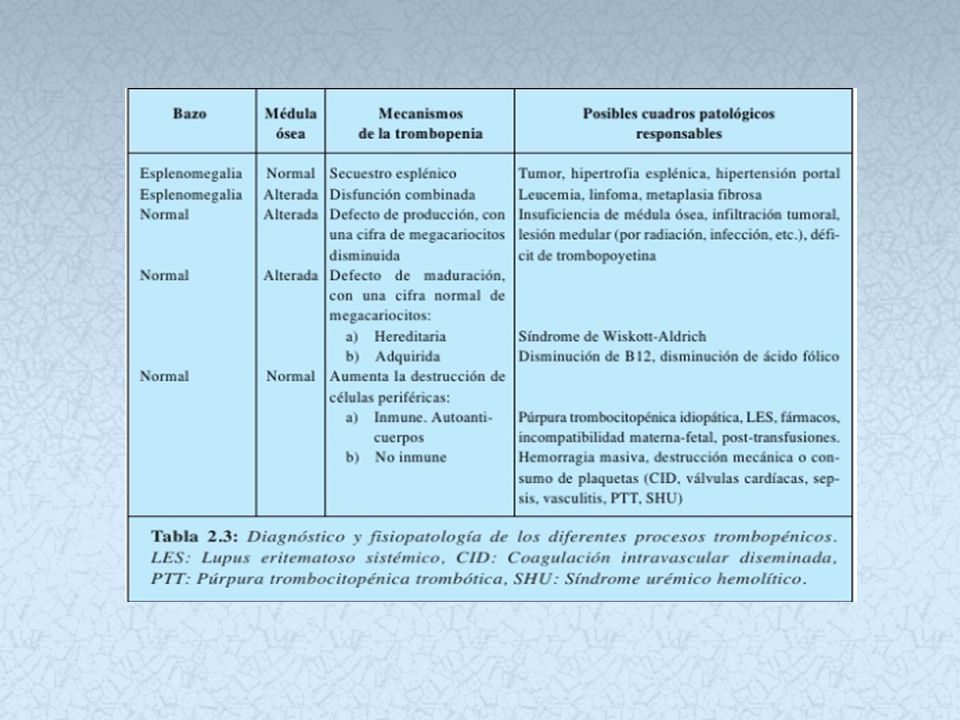
Trombocitopenia central
ETIOLOGIA: Aplasia medular Infiltración medular por cancer Infección medular por VIH ( supresión de producción de megacariocitos) Fármacos y radioterapia
 Fármacos y radioterapia.")
34
Trombocitopenia periférica
ETIOLOGIA Secuestro esplénico aumentado Autoinmune; anticuerpos antiplaquetarios.( contra glicoproteinas) Destrucción mecánica ( protesis valvulares, hipertensión maligna). Consumo excesivo ( CID -PTT)
 Destrucción mecánica ( protesis valvulares, hipertensión maligna). Consumo excesivo ( CID -PTT)")
35
Enfermedad de Von Willebrand
Es el trastorno hemorrágico hereditario más común. ETIOLOGIA: Es causada por una deficiencia del factor de von Willebrand, que ayuda a las plaquetas de la sangre a amontonarse (aglutinarse) y adherirse a las paredes de los vasos sanguíneos, lo cual es necesario para la coagulación normal de la sangre. SÍNTOMAS Sangrado menstrual anormal Sangrado de las encías Hematomas Hemorragias nasales Erupción cutánea Nota: la mayoría de las mujeres con sangrado menstrual prolongado o copioso NO TIENEN enfermedad de Von Willebrand.
 y adherirse a las paredes de los vasos sanguíneos, lo cual es necesario para la coagulación normal de la sangre. SÍNTOMAS. Sangrado menstrual anormal. Sangrado de las encías. Hematomas. Hemorragias nasales. Erupción cutánea. Nota: la mayoría de las mujeres con sangrado menstrual prolongado o copioso NO TIENEN enfermedad de Von Willebrand.")
36
Hemofilia Es una enfermedad hereditaria caracterizada por la aparición de hemorragias internas y externas debido a la deficiencia total o parcial de una proteína coagulante denominada globulina antihemofílica (factor de coagulación). Cuando hay carencia o déficit de algún factor de coagulación, la sangre tarda más tiempo en formar el coágulo y, aunque llegue a formarse, no es consistente y no se forma un buen tapón para detener la hemorragia.
. Cuando hay carencia o déficit de algún factor de coagulación, la sangre tarda más tiempo en formar el coágulo y, aunque llegue a formarse, no es consistente y no se forma un buen tapón para detener la hemorragia.")
37
Hemofilia Es una enfermedad genética que se transmite de padres a hijos. El análisis de los árboles familiares o genealogías en familias con afectados de hemofilia A o B, demostró que esta enfermedad sólo se manifestaba en los varones relacionados por vía materna y que, por tanto, debería consistir en una anomalía hereditaria que se producía por algún defecto en el cromosoma X (herencia ligada al sexo). Los cromosomas contienen las instrucciones necesarias para ordenar a las células cómo fabricar las proteínas que el organismo requiere para su funcionamiento. Estas instrucciones se encuentran contenidas en pequeñas formaciones que se llaman genes, constituidos de ADN y son la estructura básica de la vida. En cada célula hay 46 cromosomas: la mitad la recibimos como herencia de la madre y la otra mitad del padre.
. Los cromosomas contienen las instrucciones necesarias para ordenar a las células cómo fabricar las proteínas que el organismo requiere para su funcionamiento. Estas instrucciones se encuentran contenidas en pequeñas formaciones que se llaman genes, constituidos de ADN y son la estructura básica de la vida. En cada célula hay 46 cromosomas: la mitad la recibimos como herencia de la madre y la otra mitad del padre.")
38
Hemofilia La incidencia de esta enfermedad es:
Aproximadamente de forma hereditaria en un 60%. Por mutación de tipo genético y sin antecedentes en las familias en un 40%. En la mayoría de los casos hay antecedentes hereditarios conocidos en la familia. Los varones heredan la hemofilia de sus madres, aunque ellas generalmente no la padecen y en muchas ocasiones no saben que son portadoras. La mujer se comportará como portadora de la enfermedad y el hombre la padece (y transmite a la descendencia). La mujer para manifestar la enfermedad necesitaría dos copias defectuosas, cosa muy poco probable. En el caso de hija hemofílica es una situación mortal.
. La mujer para manifestar la enfermedad necesitaría dos copias defectuosas, cosa muy poco probable. En el caso de hija hemofílica es una situación mortal.")
39
HEMOFILIA - Clasificación
Hay dos variedades de hemofilia: Hemofilia A: hay un déficit del factor VIII de coagulación. Hemofilia B: hay un déficit del factor IX de coagulación.

40
Hemofilia - Síntomas El principal síntoma es la hemorragia, que puede ser externa o interna, provocada o espontánea. Las hemorragias más graves son las que se producen en: Cerebro. Ojo. Articulaciones. La hemartrosis es la manifestación clínica más frecuente en los hemofílicos. Sólo se afectan las articulaciones de un solo eje como la rodilla, el codo o el tobillo. Lengua. Garganta. Riñones. Hemorragias digestivas y genitales.

41
COAGULOPATÍA Enfermedad que consiste en un trastorno del sistema de la coagulación que funciona deficientemente. Congénitas: hemofilia o la enfermedad de Von Willebrand. Adquiridas: falta de síntesis de factores de coagulación; presencia de anticoagulantes circulantes; exceso de consumo de factores o hiperdestrucción) o en exceso (trombosis e hipercoagulabilidad).
 o en exceso (trombosis e hipercoagulabilidad).")
42
Cascada de la Agresión RESPUESTA LOCAL Fase I
TRAUMA RESPUESTA LOCAL Citocinas Macrófagos Células endoteliales Fase I RESPUESTA PARACRINA / AUTOCRINA ALTERACION EN HOMEOSTASIS Fase II SIRS Fase III Endocrina Cerebro Hematológica Hígado Corazón Pulmón Renal Intestino Metabólico MODS/MOFS

43
DESARROLLO DE LAS ALTERACIONES DE LA COAGULACIÓN
III-1 Coagulation Cascade Abnormalities (García-de-Lorenzo) DESARROLLO DE LAS ALTERACIONES DE LA COAGULACIÓN activación de la coagulación Situaciones que conducen a la trombosis oclusión trombótica de la microcirculación en todos los órganos fibrinolisis en la microcirculación consumo de plaquetas y de las proteínas de la coagulación productos circulantes de la degradación de la fibrina Boldt J. Changes of the hemostatic network in the critically ill. In Yearbook of Intensive Care and Emergency Medicine (Vincent JL, ed.) Springer-Verlag, Berlin; signos de trombosis microvascular signos de diátesis hemorrágica Boldt J. In Yearbook of Intensive Care and Emergency Medicine 2001
 DESARROLLO DE LAS ALTERACIONES DE LA COAGULACIÓN. activación de la coagulación. Situaciones que. conducen a la trombosis. oclusión trombótica de la microcirculación en todos los órganos. fibrinolisis en la microcirculación. consumo de plaquetas y de las proteínas de la coagulación. productos circulantes de la degradación de la fibrina. Boldt J. Changes of the hemostatic network in the critically ill. In Yearbook of Intensive Care and Emergency Medicine (Vincent JL, ed.) Springer-Verlag, Berlin; signos de trombosis microvascular. signos de diátesis hemorrágica. Boldt J. In Yearbook of Intensive Care and Emergency Medicine")
44
Degradación de la Fibrina
Fisiopatología t-PA u-PA XIIa Plasminógeno Plasmina Degradación de la Fibrina PAI-1 C1-Inhibidor α2-antiplasmina Plasminógeno bajo Consumo Degradación PAI-1 elevado TAT >> PAP TAT >> PAP: complejo trombina-antitrombina mas elevado que complejo plasmina-antiplasmina

45
Paso IV: Alteración Plaquetar
Trombocitopenia por destrucción microvascular y secuestro en diferentes órganos (hígado, pulmón, intestino). Aumento en la agregación y adhesividad plaquetar por afectación de la expresión de las moléculas de superficie y producción disminuida del tromboxano A2 plaquetar.
. Aumento en la agregación y adhesividad plaquetar por afectación de la expresión de las moléculas de superficie y producción disminuida del tromboxano A2 plaquetar.")
46
Resumen Los disbalances del sistema homeostático observados en el paciente crítico, son habitualmente secundarios a un sangrado excesivo o a una activación múltiple de las vías inflamatorias. La activación de mecanismos pro-coagulantes y la retro-regulación de las vías anti-coagulantes también contribuyen a ese disbalance.

47
Resumen Activación sistémica e intravascular de la coagulación
Amplios depósitos de fibrina Coagulopatía de consumo (sangrado) Fallo orgánico (isquémico) Activación del sistema de coagulación Depósitos de fibrina Coagulopatía de consumo Trombosis microvascular Sangrado (severo)
 Fallo orgánico (isquémico) Activación del sistema de coagulación. Depósitos de fibrina. Coagulopatía de consumo. Trombosis. microvascular. Sangrado (severo)")
48
Implicaciones Pronósticas
III-1 Coagulation Cascade Abnormalities (García-de-Lorenzo) Implicaciones Pronósticas La coagulopatía, cuando se presenta con hipotermia y acidosis metabólica, se asocia a elevada mortalidad. La coagulopatía es la causa más común de sangrado-relacionado con la mortalidad en el periodo post-operatorio precoz. Hoyt DB, Bulger EM, Knudson MM, et al. Death in the operating room: an analysis of a multi-center experience. J Trauma 1994; 37: Hoyt DB et al. J Trauma 1994; 37: 426
 Implicaciones Pronósticas. La coagulopatía, cuando se presenta con hipotermia y acidosis metabólica, se asocia a elevada mortalidad. La coagulopatía es la causa más común de sangrado-relacionado con la mortalidad en el periodo post-operatorio precoz. Hoyt DB, Bulger EM, Knudson MM, et al. Death in the operating room: an analysis of a multi-center experience. J Trauma 1994; 37: Hoyt DB et al. J Trauma 1994; 37: 426.")
Presentaciones similares
































 es una alteración fisiopatólogica sistémica, trombohemorrágica, que se presenta en algunas situaciones.>")







