Descargar la presentación
La descarga está en progreso. Por favor, espere

1
Laboratorio de Genética Molecular de la Hemofilia
TECNICAS DE BIOLOGIA MOLECULAR Dra Liliana Rossetti Laboratorio de Genética Molecular de la Hemofilia Cátedra de Genética y Biología Molecular. Facultad de Farmacia y Bioquímica.UBA

2
GENERALIDADES DEL GENOMA
Diploide: 2x pb= genes No codificante Codificante 95-97 % Sec intergénicas e intragénicas 3-5 % 99,9 % secuencias compartidas 0,1 % secuencias diferentes Pb 99,9% del genoma total son secuencias COMPARTIDAS por los individuos 0,1% del genoma total DIFIERE entre los individuos POLIMORFISMOS genes en genoma humano

3
gen eucariota CAAT ATG TAA/TAG/TGA 5´ 3´ gt ag E1 E2 i1 ATA AATAAA

4
E1 E2 E3 i1 i2 5´ 3´ transcripto primario capping 5´ splicing
poli A 3´ ARNm NH2 COOH ß GLOBINA

5
Extracción y purificación de ADN
Ahora....como separo el DNA? (Tejidos, cultivos celulares, sangre) Obtención de la muestra PRECIPITACION - Sales (apantallan cargas) - Alcohol (dism. Cte dielectrica) RESUSPENDER - H2O - TE Separar las células de interés Disgregar y homogeneizar (sin romper las células!!!) ROMPER LAS CELULAS!!! - Medios hipotónicos - Sonicado - detergentes (FL) - Enzimas (Proteínas) INHIBICION DE DNAsas!! Fase acuosa enriquecida en ADN EXTRACCION - Sv. Orgánicos (Fenol, cloroformo, eter) CHEQUEO DEL ADN -Espectrofotometria - Electroforesis Extracción y purificación de ADN
 Obtención de la muestra. PRECIPITACION. - Sales (apantallan cargas) - Alcohol (dism. Cte dielectrica) RESUSPENDER. - H2O. - TE. Separar las células de interés. Disgregar y homogeneizar (sin romper las células!!!) ROMPER LAS CELULAS!!! - Medios hipotónicos. - Sonicado. - detergentes (FL) - Enzimas (Proteínas) INHIBICION DE DNAsas!! Fase acuosa enriquecida en ADN. EXTRACCION. - Sv. Orgánicos. (Fenol, cloroformo, eter) CHEQUEO DEL ADN. -Espectrofotometria. - Electroforesis. Extracción y purificación de ADN.")
6
Chequeo del ADN obtenido
ELECTROFORESIS INTEGRIDAD CANTIDAD Cc conocida (-) (+) (-) (+) Matriz semisólida: GEL de AGAROSA Visualización al UV: Bromuro de Etidio Se corre a pH básico o neutro ADN carga (-) MIGRA AL POLO (+)
 (+) (-) (+) Matriz semisólida: GEL de AGAROSA. Visualización al UV: Bromuro de Etidio. Se corre a pH básico o neutro ADN carga (-) MIGRA AL POLO (+)")
7
Extracción y purificación de ARN
(Tejidos, cultivos celulares, sangre, MO) Obtención de la muestra Separar las células de interés Disgregar y homogeneizar (sin romper las células!!!) Ahora....como separo el RNA? ROMPER LAS CELULAS!!! TRIZOL Aislar RNA en un solo paso Tiocianato de guanidinio/fenol INHIBICION DE RNAsas!! EXTRACCION - Sv. Orgánicos Cloroformo PRECIPITACION - Alcohol (dism. Cte dielectrica) Fase acuosa enriquecida en ARN RESUSPENDER - H2O - TE CHEQUEO DEL ARN -Espectrofotometria - Electroforesis
 Obtención de la muestra. Separar las células de interés. Disgregar y homogeneizar (sin romper las células!!!) Ahora....como separo el RNA ROMPER LAS CELULAS!!! TRIZOL. Aislar RNA en. un solo paso. Tiocianato de. guanidinio/fenol. INHIBICION DE RNAsas!! EXTRACCION. - Sv. Orgánicos. Cloroformo. PRECIPITACION. - Alcohol (dism. Cte dielectrica) Fase acuosa enriquecida en ARN. RESUSPENDER. - H2O. - TE. CHEQUEO DEL ARN. -Espectrofotometria. - Electroforesis.")
8
Chequeo del ARN obtenido
ELECTROFORESIS INTEGRIDAD CANTIDAD Matriz semisólida: GEL de AGAROSA Visualización al UV: Bromuro de Etidio (-) (+) (-) (+) Se corre a pH básico o neutro, ARN carga (-) MIGRA AL POLO (+)
 (+) (-) (+) Se corre a pH básico o neutro, ARN carga (-) MIGRA AL POLO (+)")
9
Chequeo del ADN obtenido
ESPECTROFOTOMETRÍA Pureza Relación 260nm / 280nm 1,8-2 Concentración 1D.O. = 50 µg/ml ADN dc 40 µg/ml ADN sc o ARN 20 µg/ml oligonucleótidos

10
CORRIDA ELECTROFORETICA
PREPARADO DEL GEL SIEMBRA DE LA MUESTRA CORRIDA ELECTROFORETICA Muestra Buffer de siembra (ej: ABF) densidad marca frente de corrida Agarosa Buffer de corrida (ej:TBE) Poliacrilamida Buffer de corrida (ej:TBE) Corrida en agarosa ADN cortado con ER Corrida en PAGE Prod PCR tincion con Ag
 densidad. marca frente de corrida. Agarosa. Buffer de corrida (ej:TBE) Poliacrilamida. Buffer de corrida (ej:TBE) Corrida en agarosa. ADN cortado con ER. Corrida en PAGE. Prod PCR tincion con Ag.")
11
A igual carga neta Diferencia por PM
Cómo discrimina un gel? A igual carga neta Diferencia por PM A MAYOR PM MENOR MIGRACION 1000 pb 2000 pb 3000 pb 4000 pb 5000 pb 6000 pb 7000 pb 8000 pb 9000 pb 10000 pb Log PM Dist Interpolo fragmento de PM desconocido Marker de 1Kb

12
Cuánto discrimina un gel?
Agarosa 2% Poliacrilamida 6%

14
SOUTHERN BLOT Electroforesis en Purificación del ADN gel de agarosa
Papel Gel de agarosa Membrana de nylon Electroforesis en gel de agarosa Purificación del ADN Transferencia Radioautografía Hibridación Marcación de la sonda

15
Análisis mediante Southern blot de deleciones en los genes de -globina
Enzima de restricción: BamH I : 14Kb -3.7:10,3Kb ELECTROFORESIS AUTORRADIOGRAFIA 23,13 Kb 9,41 Kb 6,56 Kb 4,36 Kb 2,32 Kb 2,03 Kb 14,0 Kb 10,3 Kb

17
PCR Reacción en Cadena de la Polimerasa
Definición: amplificación enzimática in vitro de una secuencia específica de ADN mediante ciclos repetidos de desnaturalización térmica, unión complementaria (annealing) de primers que definen la zona de interés y elongación de los mismos, catalizada por una enzima termoestable (Taq polimerasa). Características: sensible y específica.
 de primers que definen la zona de interés y elongación de los mismos, catalizada por una enzima termoestable (Taq polimerasa). Características: sensible y específica.")
18
PCR Reacción en Cadena de la Polimerasa
Ciclado programable Enzima polimerizante termoestable (Taq, Pfu,etc.) permiten aumentar exponencialmente la cantidad de ADN flanqueado por un par de primers.
 permiten aumentar exponencialmente la cantidad de ADN flanqueado por un par de primers.")
19
Desnaturalización Annealing
5’ 3’ 5’ 3’ Desnaturalización 5’ 3’ Annealing 5’ 3’ 5’ 3’ 5’ 3’ Polimerización Taq pol dNTPs 3’ 5’ 5’ 3’

20
Annealing y Polimerización
5’ 3’ Desnaturalización 5’ 3’ 5’ 3’ Annealing y Polimerización fragmentos limitados por primers

21
2do ciclo 3er ciclo 5’ 3’ 3’ 5’ 3’ 5’ 5’ 3’ 3’ 5’ 5’ 3’ 3’ 5’ 5’ 3’ 3’

22
Segundo ciclo Primer ciclo 94 °C 94 °C Polimerización 72 °C 72 °C
Desnaturalización 94 °C 94 °C Polimerización 72 °C 72 °C °C °C Annealing Primer ciclo Segundo ciclo

23
En el tercer ciclo se completa la primera copia del producto
AMPLIFICACION En el segundo ciclo se determina la longitud del producto. En el tercer ciclo se completa la primera copia del producto

24
REACTIVOS Templado: ADNdc 1 ng – 50 ng
Primers (sense-antisense): 50 pmol c/u Taq ADN polimerasa: 1U Buffer 10x Cl2Mg: 1-5 mM dNTPs (x4): 200 µM c/u H2O: csp 50 µl * Factores controlables
: 50 pmol c/u. Taq ADN polimerasa: 1U. Buffer 10x. Cl2Mg: 1-5 mM. dNTPs (x4): 200 µM c/u. H2O: csp 50 µl. * Factores controlables.")
25
PCR Reacción en Cadena de la Polimerasa
Temperatura de annealing. Longitud y secuencia de los primers. Concentración de primers. Concentración de Cl2Mg.

27
* These sequences have the H274Y mutation that confers resistance to Oseltamivir.

28
Diseño de primers

32
PCR directa para detectar un polimorfismo de longitud
Alu Pv92

33
GENERALIDADES DEL GENOMA
Diploide: 2x pb= genes No codificante Codificante 95-97 % Sec intergénicas e intragénicas 3-5 % 99,9 % secuencias compartidas 0,1 % secuencias diferentes Pb 99,9% del genoma total son secuencias COMPARTIDAS por los individuos 0,1% del genoma total DIFIERE entre los individuos POLIMORFISMOS genes en genoma humano

34
Polimorfismos Tipos de polimorfismos
Variación en una secuencia nucleotídica presente en una población con una frecuencia mayor al 1% Generalmente se atribuyen a secuencias que no generan patología Tipos de polimorfismos Polimorfismo de secuencia Polimorfismo de longitud VNTR´S: polimorfismos de repetición Microsatélites o STR´s core (2-7pb) n = 1 a 20 repeticiones Minisatélites:core (5- 100pb) n aprox 1 a 100 repeticiones SNP´s: variación de un único nucleótido Si modifica el sitio de corte de una enzima : RFLP
 n = 1 a 20 repeticiones. Minisatélites:core (5- 100pb) n aprox 1 a 100 repeticiones. SNP´s: variación de un único nucleótido. Si modifica el sitio de corte de una enzima : RFLP.")
35
Polimorfismos (ej: SNP)
5´ACGGTACGATACGTAGGGCTAGGCTACTAGGAT 3´ 5´ACGGTACGATACGTAGGGCTAGGCTACTAGGAT 3´ 5´ACGGTACGATACGTAGGGCTAGGCTACTAGGAT 3´ 5´ACGGTACGATACGTAGGGCTAGGCTACTAGGAT 3´ 5´ACGGTATGATACGTAGGGCTAGGCTACTAGGAT 3´ 5´ACGGTACGATACGTAGGGCTAGGCTACTAGGAT 3´ 5´ACGGTACGATACGTAGGGCTAGGCTACTAGGAT 3´ 5´ACGGTACGATACGTAGGGCTAGGCTACTAGGAT 3´ 5´ACGGTACGATACGTAGGGCTAGGCTACTAGGAT 3´ 5´ACGGTACGATACGTAGGGCTAGGCTACTAGGAT 3´ Cuantas variantes alélicas dif hay en esta población? 2= C / T Cuantos genotipos diferentes hay en esta población? 2= (C/C) (C/T) 4= homo 1 = hetero Cuantos Homo y heterocigotas?
 (C/T) 4= homo 1 = hetero. Cuantos Homo y heterocigotas")
36
Polimorfismos ej:microsatélite
5´ACGGTACACAGATACGTAGGGCTAGGCTACTAGGAT 3´ 5´ACGGTACACAGATACGTAGGGCTAGGCTACTAGGAT 3´ 5´ACGGTACAGATACGTAGGGCTAGGCTACTAGGAT 3´ 5´ACGGTACACACAGATACGTAGGGCTAGGCTACTAGGAT 3´ 5´ACGGTACACACAGATACGTAGGGCTAGGCTACTAGGAT 3´ 5´ACGGTACACAGATACGTAGGGCTAGGCTACTAGGAT 3´ 5´ACGGTACACACACAGATACGTAGGGCTAGGCTACTAGGAT 3´ 5´ACGGTACAGATACGTAGGGCTAGGCTACTAGGAT 3´ 5´ACGGTACACAGATACGTAGGGCTAGGCTACTAGGAT 3´ 5´ACGGTACAGATACGTAGGGCTAGGCTACTAGGAT 3´ Cuantas variantes alélicas dif hay en esta población? 4= repet. Cuantos genotipos diferentes hay en esta población? 5= (2/2) (1/3) (3/2) (4/1) (2/1) Cuantos Homo y heterocigotas? 4= hetero 1 = homo
 (1/3) (3/2) (4/1) (2/1) Cuantos Homo y heterocigotas 4= hetero 1 = homo.")
37
Polimorfismos como herramienta molecular:
Filiación Evolución Forense Mapeo de genes Diagnóstico de Patologías Hereditarias (como marcador) Microsatélites involucrados en Patologías Predisposición a malignización de cánceres
 Microsatélites involucrados en Patologías. Predisposición a malignización de cánceres.")
38
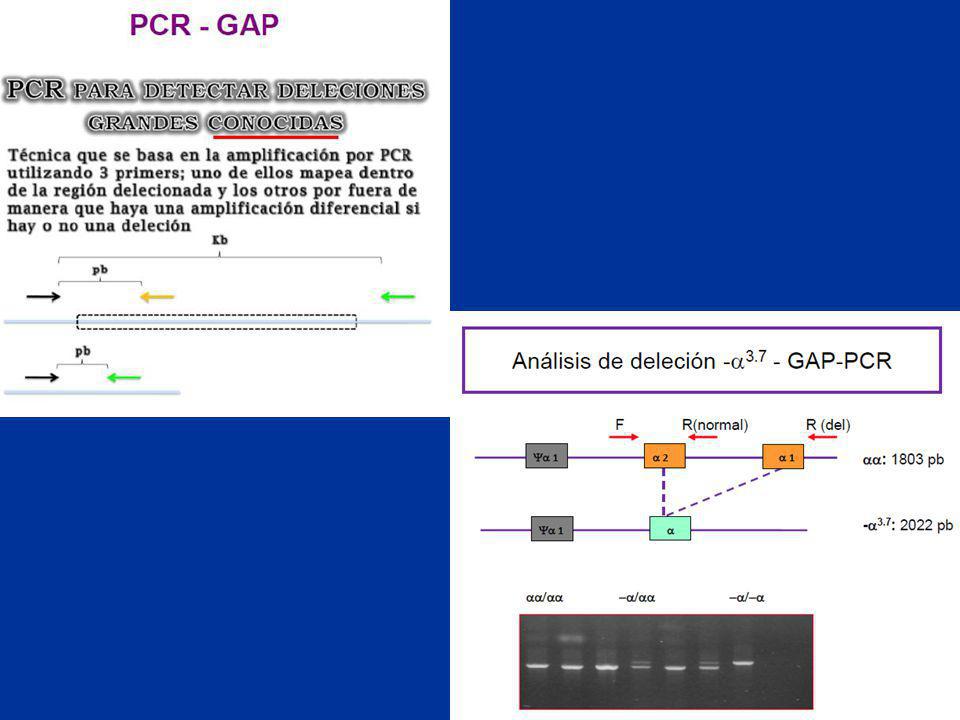
RFLP Restriction Fragment Length Polymorphisms
Definición: variaciones detectables en la longitud de los fragmentos generados al cortar DNA con una enzima de restricción. Causa: cambio en la secuencia del sitio de restricción.

39
endonucleasa de restricción
Genotipos alelo A: 11,0 y 3,5 Kb alelo B: 14,5 Kb AA BB AB Taq I exón 5’ 3’

40
El polimorfismo esta asociado al gen: se hereda en bloque.
Diagnóstico de Patologías Hereditarias Concepto de Marcador KM 19 Pst I El polimorfismo esta asociado al gen: se hereda en bloque.

41
Microsatélites (STRs)
Distribuídos a lo largo de todo el genoma Altamente polimórficos (muchas variantes alélicas) Alto nro de individuos hererocigotas gtagtagccgtagagttgc(cacacacacaca)ttcattattacattaca (ca)n n=6 Locus= lugar determinado en el genoma Alelo: secuencia de bases en un locus
 Alto nro de individuos hererocigotas. gtagtagccgtagagttgc(cacacacacaca)ttcattattacattaca. (ca)n n=6. Locus= lugar determinado en el genoma. Alelo: secuencia de bases en un locus.")
42
Electroforesis capilar automatizada

43
Electroforesis capilar

44
Microsatélites (STR’S)
Ej: agttgc(cacacacacaca)ttcatt agttgc(cacaca)ttcatt agttgc(cacacaca)ttcatt agttgc(caca)ttcatt haplotipo 6 4 3 2 agttgc(cacacacacaca)ttcatt 6 2 agttgc(caca)ttcatt
ttcatt. agttgc(cacaca)ttcatt. agttgc(cacacaca)ttcatt. agttgc(caca)ttcatt. haplotipo agttgc(cacacacacaca)ttcatt agttgc(caca)ttcatt.")
45
Filiación padre 1? padre 2? ?

46
Filiación padre 1? padre 2? inclusión exclusión

47
Sistema de identificación genética
PowerPlex® 16 System

48
Filiación padre 1? padre 2? inclusión exclusión

49
Forense / criminalística
Sospechoso 1 Sospechoso 2 inclusión exclusión

50
CDNA distrofina exón DMB DMD 79 78 77 76 75 74 73 72 71 70 69 68 67 66 65 64 63 62 61 60 59 58 57 56 55 54 53 52 51 50 49 48 47 46 45 44 43 42 41 40 39 38 37 36 35 34 33 32 31 30 29 28 27 26 25 24 23 22 21 20 19 18 17 16 15 14 13 12 11 10 9 8 7 6 5 4 3 2 1 32 % 62 % Ubicación de las deleciones de los pacientes con DMD/DMB en el gen de la distrofina.

51
Chequeo de los productos de PCR en gel de agarosa 2%
PCR MULTIPLEX 4 cuadruplex: 4 pares de primers cada una = 16 exones Se estudiaron las zonas más propensas a sufrir deleciones en el gen de la distrofina (Mix I, Mix II, Mix III y Mix IV). 90 % de las deleciones Mpm 3 4 8 13 17 19 43 44 45 47 48 50 51 52 60 5´ 3´ PCR: ADN primers buffer de la enz Cl2Mg dntps agua enzima (TAQ pol) Chequeo de los productos de PCR en gel de agarosa 2% Br de Et MIXI MIXII MIXIII MIXIV
. 90 % de las deleciones. Mpm ´ 3´ PCR: ADN. primers. buffer de la enz. Cl2Mg. dntps. agua. enzima (TAQ pol) Chequeo de los productos de PCR en gel de agarosa 2% Br de Et. MIXI. MIXII. MIXIII. MIXIV.")
52
Resultados 406 MIX I + - MIX II EXON 4 8 (506pb) EXON 17 (416 pb)
MIX IV 406 407 - 408B + EXON 43 (357 pb ) EXON 60 (139 EXON 47 (181 EXON 50 (271 MIX III 406 + - EXON 3 (410 pb ) Mpm (535 EXON 4 (196 EXON 13 (238
 EXON 60 (139. EXON 47 (181. EXON 50 (271. MIX III EXON 3 (410. pb. ) Mpm. (535. EXON 4 (196. EXON 13 (238.")
54
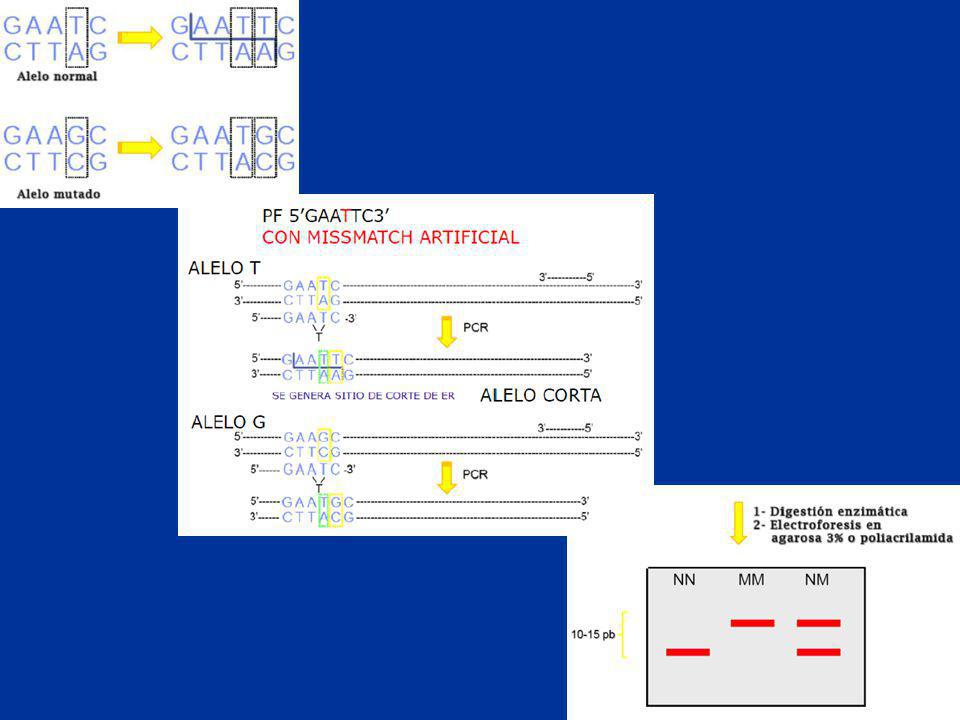
ASO Allele Specific Oligoprobe
Definición: identificación de mutaciones puntuales (sin sitio de restricción) mediante 2 sondas complementarias a los alelos N y M, con secuencia nucleotídica conocida.
 mediante 2 sondas complementarias a los alelos N y M, con secuencia nucleotídica conocida.")
55
desnaturalización alcalina siembra en membrana de nylon
Detección de mutaciones puntuales Dot-Blot e hibridación con sondas de oligonucleótidos aleloespecíficas ADN PCR desnaturalización alcalina siembra en membrana de nylon hibridación con ASO

56
ALTA ASTRINGENCIA - 100% DE HOMOLOGÍA
TECNICA PCR con primer secuencia-específicos flanqueantes al sitio de mutación. DOT BLOT: transferencia por vacío a soporte sólido. Desnaturalización (DNAsc) Hibridación con oligosondas alelo específicas-ASO- ALTA ASTRINGENCIA - 100% DE HOMOLOGÍA Autoradiografía.
 Hibridación con oligosondas alelo específicas-ASO- ALTA ASTRINGENCIA - 100% DE HOMOLOGÍA. Autoradiografía.")
57
AAGTTGTACGTTCTAGGCGT AAGTTGTACCTTCTAGGCGT
Sonda Normal Sonda Mutada AAGTTGTACGTTCTAGGCGT AAGTTGTACCTTCTAGGCGT Resultado de hibridación

62
RT-PCR

63
RT-PCR

64
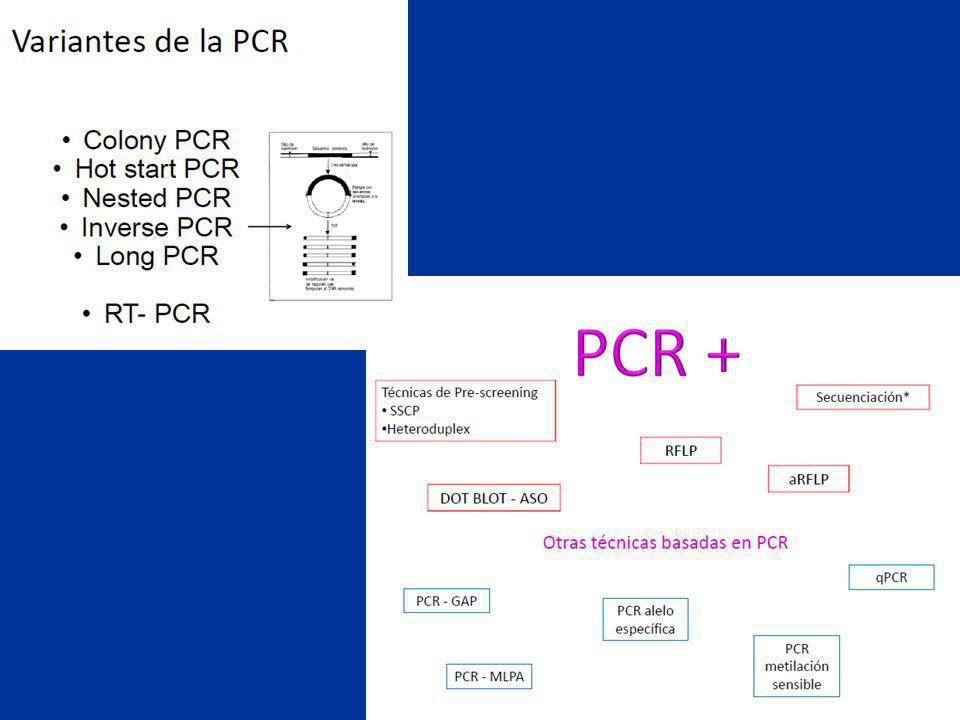
Técnicas de Prescreening

65
Estrategia condicionada por:
gen mutación caracterizado conocida caracterizado muy variable / desconocida no caracterizado desconocida indirecto directo diagnóstico

66
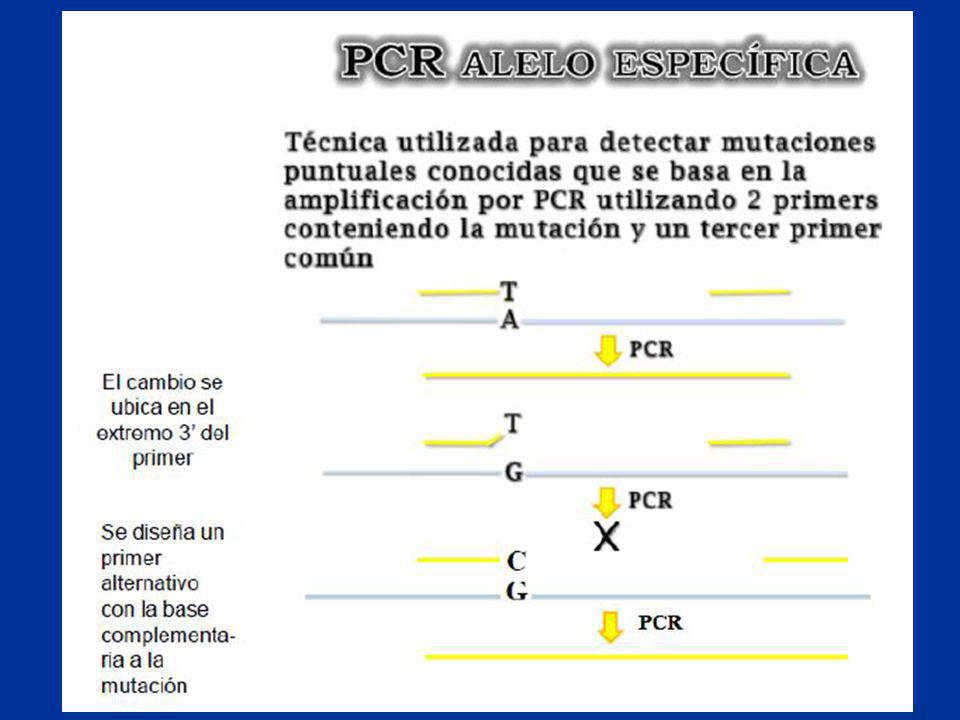
Búsqueda de mutaciones puntuales:
Prescreening SSCP/ Heteroduplex Búsqueda de mutaciones puntuales: ? 5` 3` - Permite diferenciar hasta una base - Se ven diferentes patrones de corrida

67
Técnica de heteroduplex
Prescreening Técnica de heteroduplex 1 2 3 4 Muestra a secuenciar Gel de poliacrilamida 10%-Nitrato de Plata Desnaturalización Renaturalización lenta

68
MUTACIONES IDENTIFICADAS
HETERODUPLEX DEL EXÓN 23 23 SECUENCIACIÓN G A T C G A T C C G A C T T g C>T CGA TGA Arg Stop R787X BANDA DE MIGRACIÓN ELECTROFORÉTICA ALTERADA Pérdida de funcionalidad de la proteína por terminación prematura

69
Prescreening SSCP: single strand conformation polymorphism
Búsqueda de mutaciones puntuales 5` 3` 1) Se amplifica un exón determinado por PCR ej: exón 1 en diferentes pacientes 2) Se corre en un gel en condiciones no desnaturalizantes 3) Tinción con Ag 4) Se analizan los patrones de corrida
 Se amplifica un exón determinado por PCR. ej: exón 1 en diferentes pacientes. 2) Se corre en un gel en condiciones no desnaturalizantes. 3) Tinción con Ag. 4) Se analizan los patrones de corrida.")
70
Ej: 5` 3` normal mutado Cuantas cadenas diferentes hay ?

71
Dirección de la corrida electroforética
Single Strand Conformation Polymorphism Dirección de la corrida electroforética + - (+) (-) M M
 (-) M. M.")
72
SSCP Secuenciar

73
Single Strand Conformation Polymorphism

74
Análisis de la region 5’ del gen: posición –82 a codón 91
N N M NORMAL MUTADO B T C AG T C A G 201 pb 149 pb 88-89 pb SSCP Secuenciación P5-P6/ Dde I, Gel PA 12% c/glicerol Cambio de base en el codón 30, (GC) en el gen de b-globina humana: 2 individuos
 en el gen de b-globina humana: 2 individuos.")
76
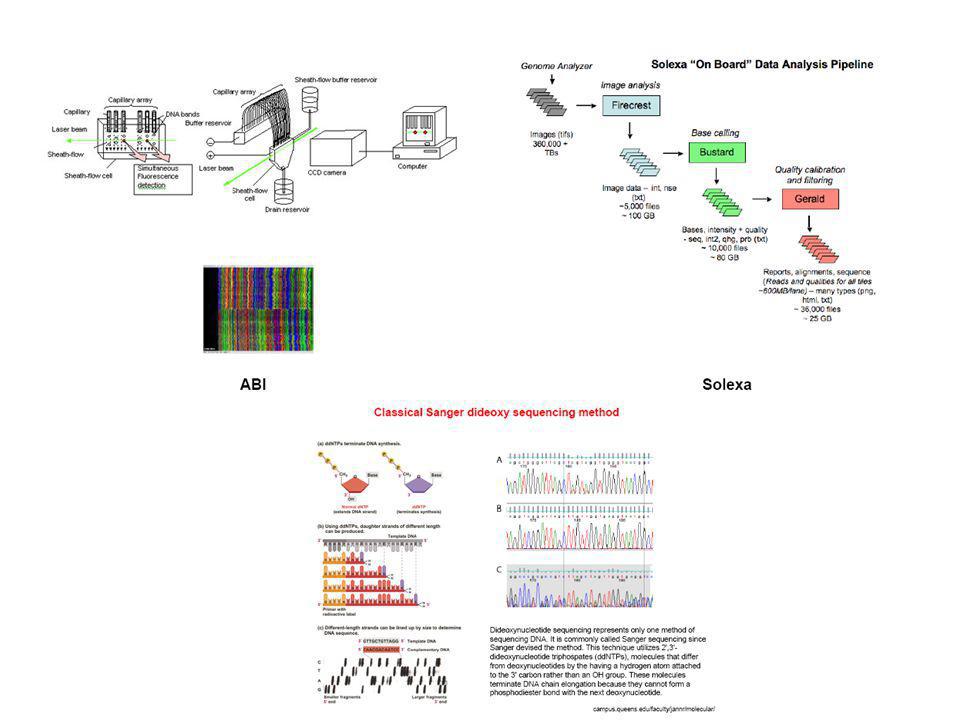
Secuenciación Def.: Determinación de la estructura primaria de los genes. Aplicaciones: Organización estructural de los genes Estudio comparativo de los genes a través de la escala evolutiva. Identificación de mutaciones Diagnóstico de enfermedades genéticas Secuenciación de ADN de fósiles Proyecto Genoma Humano Requisito: Disponer de una preparación pura y homogénea de ADN

77
Método de Sanger ADN cadena simple Cebador marcado ADN polimerasa I
3’ CGTATACAGTCAGGTC 5’ ADN cadena simple 5’ GCAT 3’ Cebador marcado ADN polimerasa I dATP dCTP dGTP dTTP + ddATP + ddTTP + ddGTP + ddCTP GCAT GCAT GCAT A GCATAT GCATATGT GCATATGTCAGT GCATATGTC GCATATGTCAGTC GCATATGTCAGTCC GCATATG GCATATGTCAG GCATATGTCAGTCCAG A T G T C A A T G T C A G T C C A A T C G TGACTGTA 5´ 3´

78
O O- P O CH2 Base O- O O O- P O CH2 Base OH H O- O dNTP H H ddNTP

79
Sanger-PCR P Dilución primer Buffer PNK PNK
ADN purificado proveniente de fago, plásmido o prod. de PCR. Buffer Taq Pol Taq Pol 30´37ºC 2´90ºC Hielo dNTP ddATP ddGTP ddCTP ddTTP P Primer marcado Ciclador térmico

80
Cubas de Electroforesis Vertical

81
1 2 3 4 5 T C A G T C A G T C AG T C A G T C A G T C g/t t a A G c/aa
Cadena Codificante T C A G T C A G T C AG T C A G T C A G T C g/t t a A G c/aa t

82
Secuenciación automática

83
Sec. Automática en gel Sec. Automática capilar

84
Sistema de detección

85
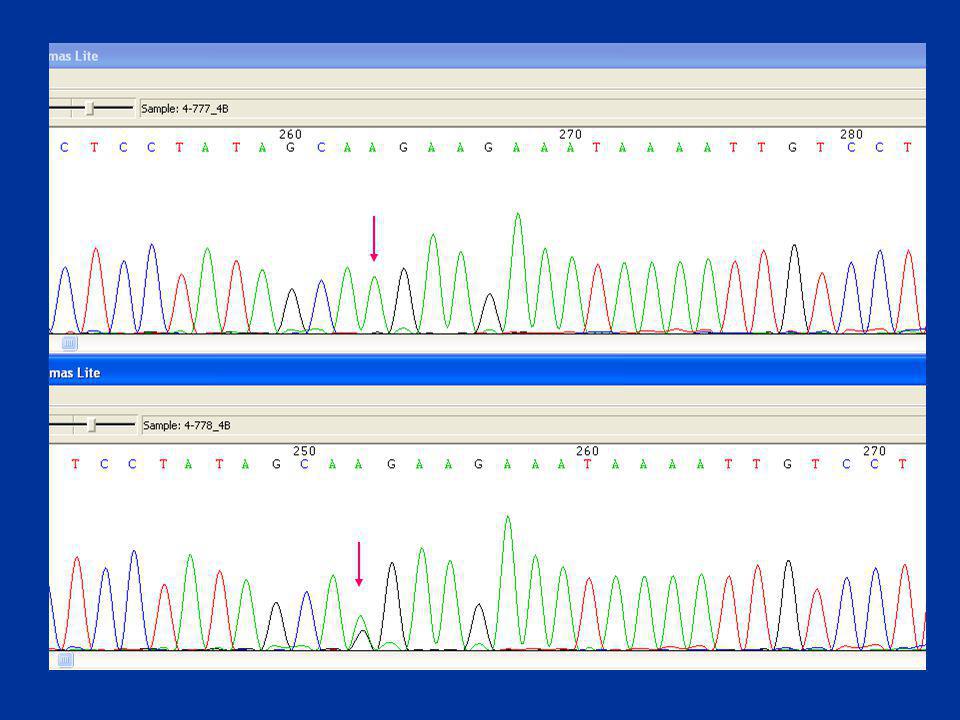
Cromatograma

86
Cromatograma

88
Secuenciación automática Exón 14E gen F8
Ref Sec. Control normal delA en un A-run; c.3637delA; p.Ile1441Phe fs+5X

89
Secuenciación automática Exón 7 gen F8
Ref Ref c.901C>T p.Arg282Cys Sec. Control normal

90
Secuenciación automática Exón 14 gen F8
insA en un A-run; c.4379_4380 insA; p.Asn1441Lys fs+2X

93
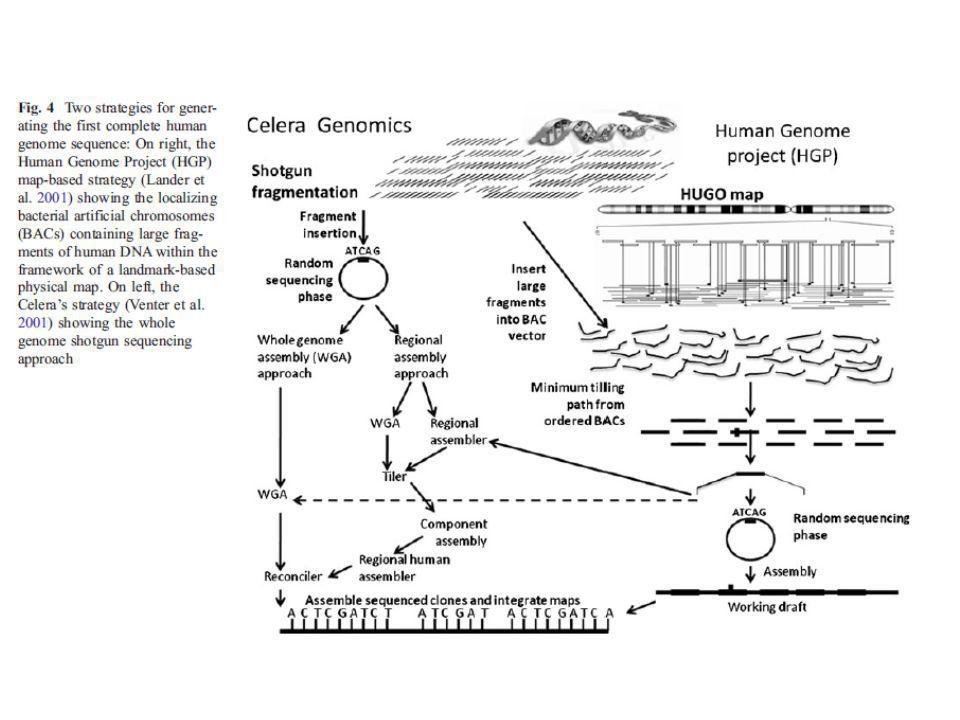
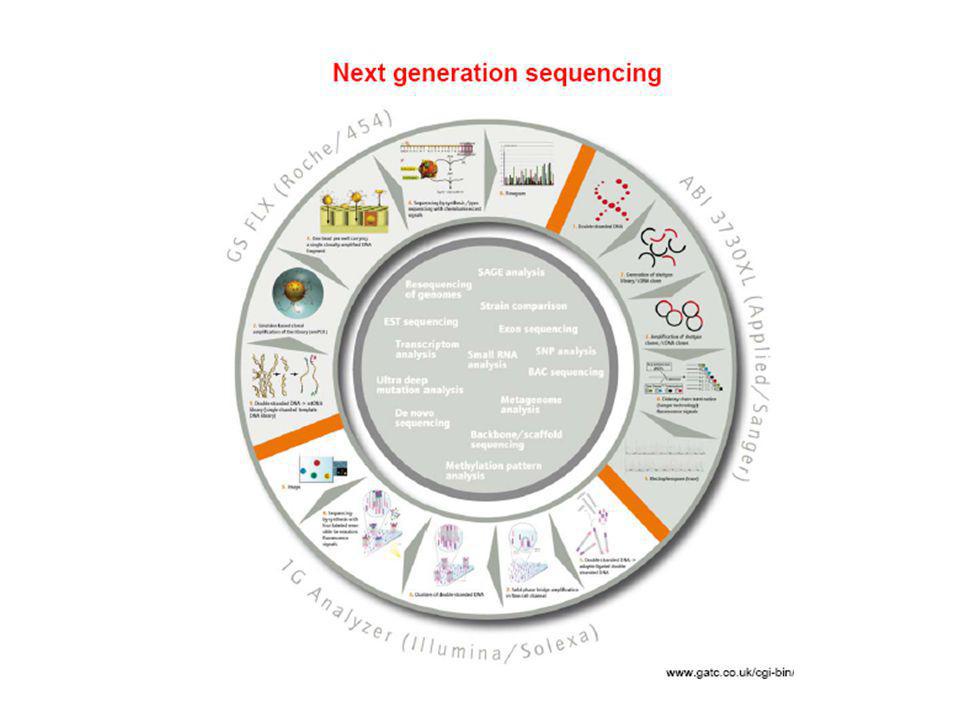
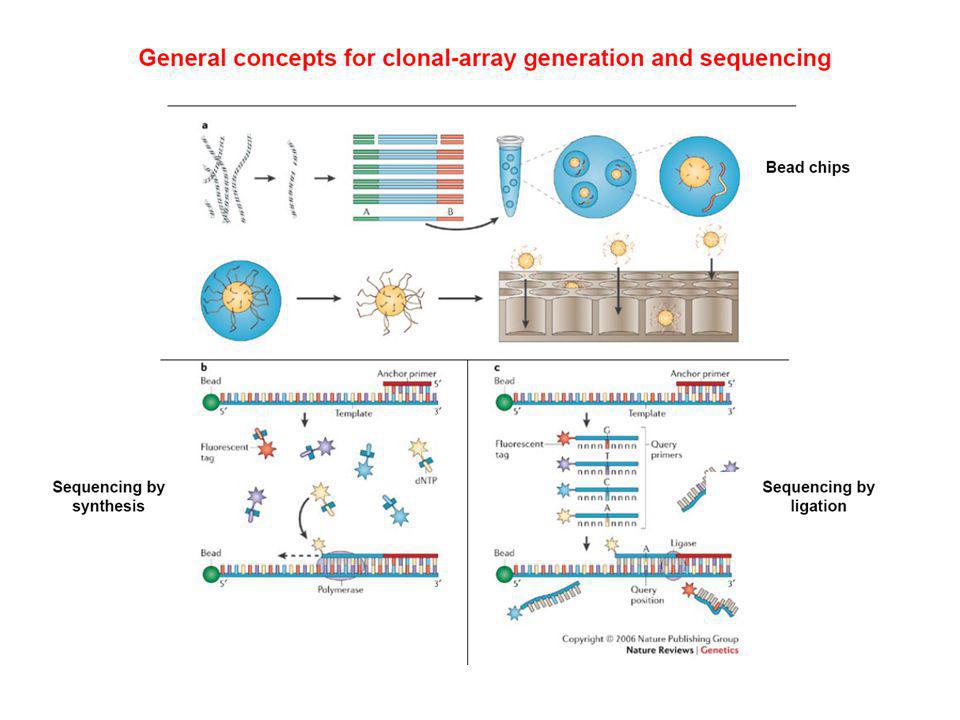
Tecnología de determinación de secuencia de DNA-RNA a gran escala.
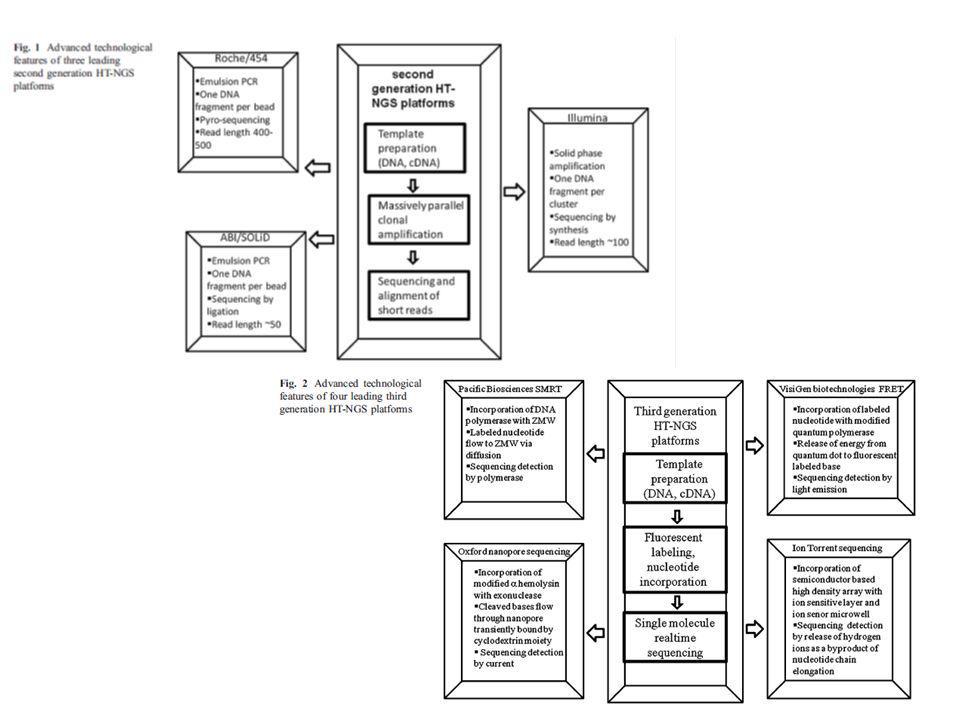
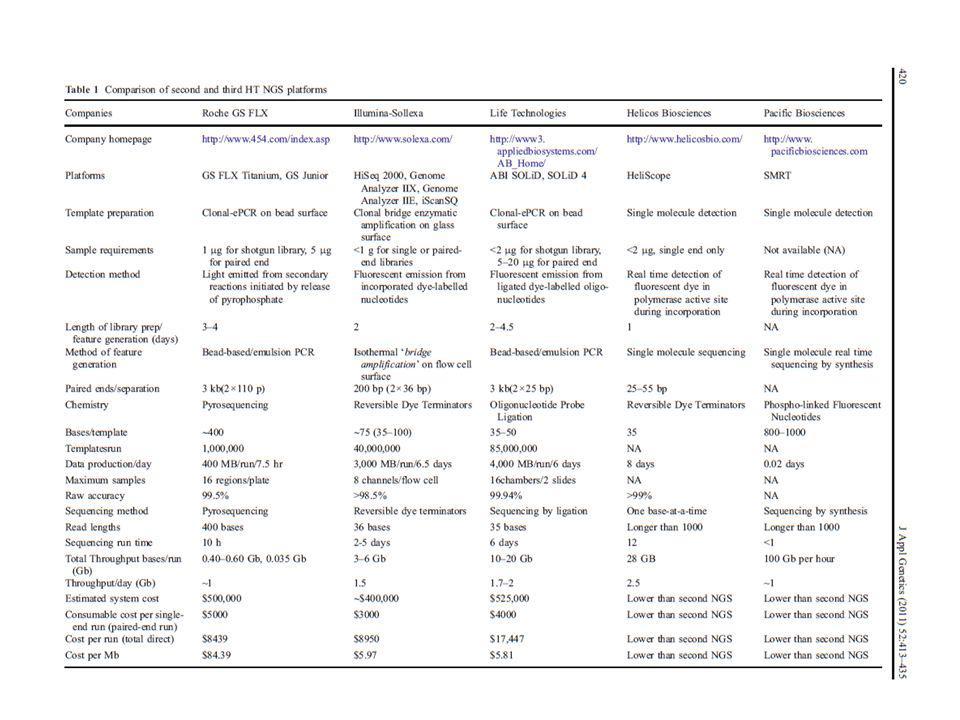
Aplicable a genomas completos mediante luminiscencia Entre las metodologías más conocidas: Secuenciación mediante amplificación con esferas (Roche/454FLX) Secuenciación por síntesis (Illumina/Solexa Genome analyzer) Secuenciación mediante ligación (Applied Biosystems SOLID System) Características comunes: Tecnología de alta complejidad, donde se conjugan diferentes tipos de enzimas, químicas, software, hardware, ingeniería óptica….. Preparación de las muestras muy eficiente previa a la secuenciación para que este paso sea mas sencillo (ahorra tiempo). Preparación de ADN de interés en librerías mediante plataformas y linkers específicos y amplificación. Amplificación de los fragmentos en cadena simple, y realización de la secuenciación sobre estos fragmentos ya amplificados
 Secuenciación por síntesis (Illumina/Solexa Genome analyzer) Secuenciación mediante ligación (Applied Biosystems SOLID System) Características comunes: Tecnología de alta complejidad, donde se conjugan diferentes tipos de enzimas, químicas, software, hardware, ingeniería óptica….. Preparación de las muestras muy eficiente previa a la secuenciación para que este paso sea mas sencillo (ahorra tiempo). Preparación de ADN de interés en librerías mediante plataformas y linkers específicos y amplificación. Amplificación de los fragmentos en cadena simple, y realización de la secuenciación sobre estos fragmentos ya amplificados.")
94
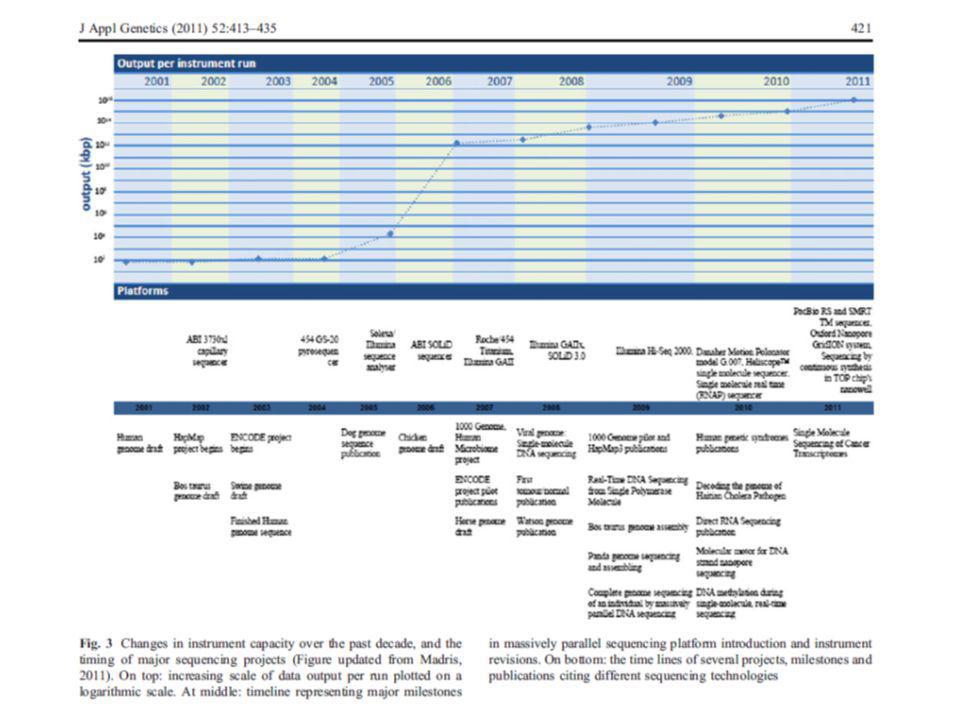
Comparación entre secuenciación de primera generación (Sanger automatizada) y secuenciación masiva (NGS: Next Generation Sequencing) - Una inmensa diferencia en el tiempo de procesamiento: una corrida en: 96 capilares de 750 pb comparada con varios miles (Roche) a millones (Illumina, ABi) de pequeñas lecturas. -Los tiempos de corrida: NGS (8h – 10days) - Sanger: bases en una hora - Roche 454: 20 millones en 4,5 horas. (abarata enormemente el coste del proceso) Costo secuenciación HGP: US$ NGS: US$ – 2nd – 3rd generation HT-NGS: US$ ?
 a millones (Illumina, ABi) de pequeñas lecturas. -Los tiempos de corrida: NGS (8h – 10days) - Sanger: bases en una hora. - Roche 454: 20 millones en 4,5 horas. (abarata enormemente el coste del proceso) Costo secuenciación HGP: US$ NGS: US$ – nd – 3rd generation HT-NGS: US$")
97
Paso 1: Obtener biblioteca de ADN simple cadena
Esta técnica comienza con el procesado y adaptación del ADN para obtener una biblioteca de pequeños fragmentos monocatenarios. Lo primero es fragmentar el ADN a secuenciar mediante un proceso físico conocido como “nebulización” donde el ADN se rompe en fragmentos de 200 a 800 pares de bases aproximadamente.

98
Paso 2: amplificacion por PCR
Paso 3: secuenciacion puede ser de dos tipos En fase SOLIDA En fase LIQUIDA Esferas magneticas o de sefarosa, revestidas con estreptovidina Enzimas degradadora de nucleotidos: aspirasa Enzima que degrada los primers remanentes el ADN esta marcado con biotina

102
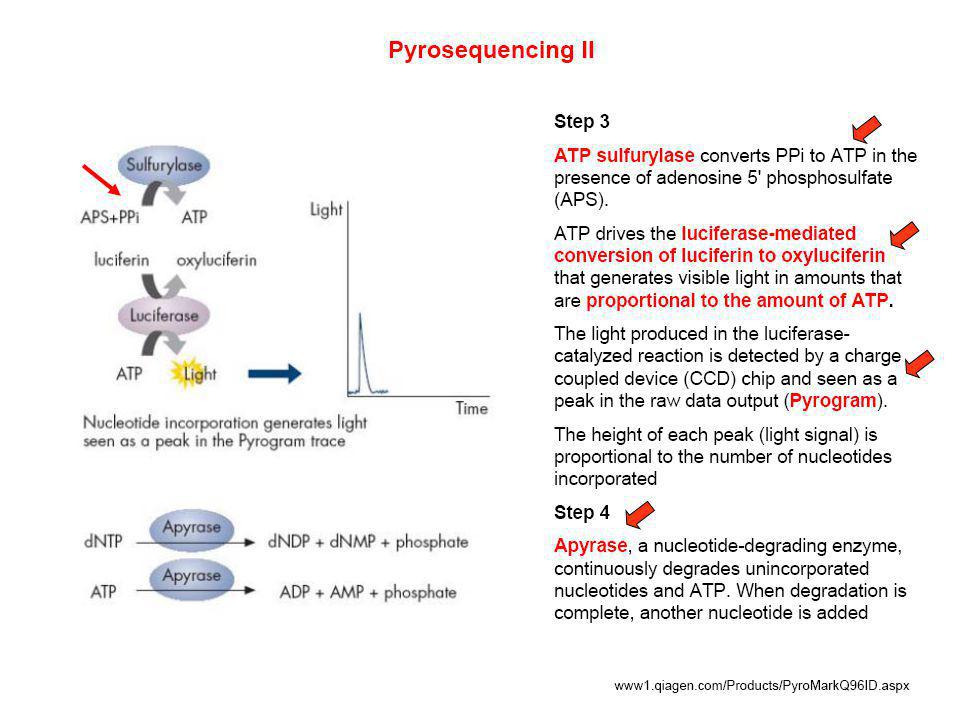
DESVENTAJA DE LA TECNICA
VENTAJAS DE LA TECNICA gran exactitud flexibilidad facilmente automatizable la luz generada es proporcional al numero de nucleotidos incorporados sin importar cual sea DESVENTAJA DE LA TECNICA el costo de los primers marcados con biotina que usualmente se usa en estos metodos. se necesita instrumentos especializados. 102

103
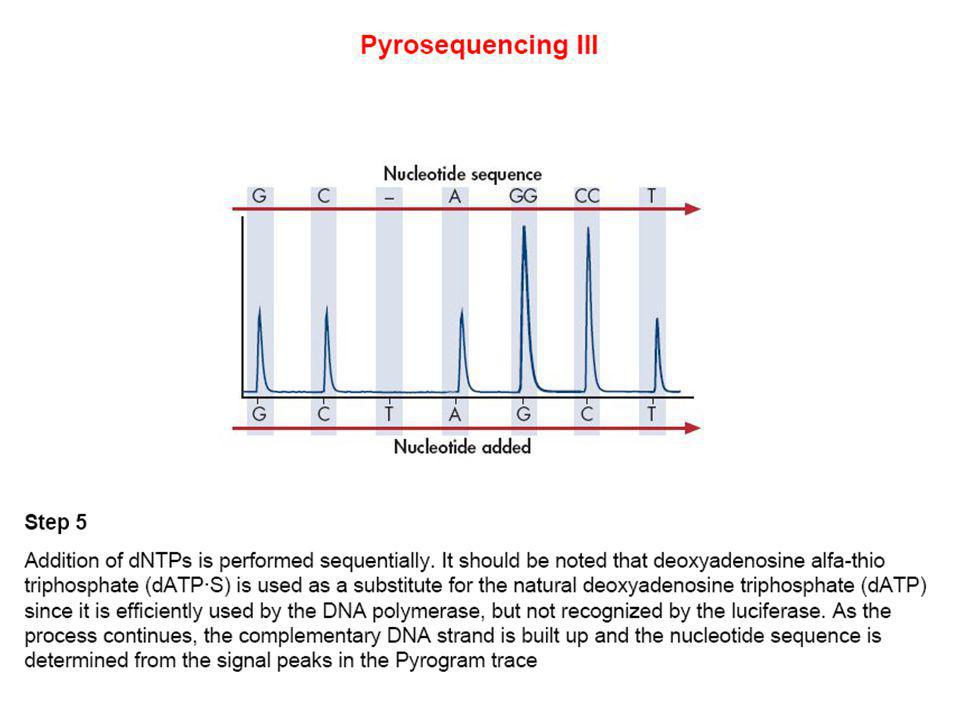
MODELO DEL EQUIPO DE PIROSECUENCIACION

104
Aplicaciones Analisis de secuencia Deteccion de mutaciones
Identificación de secuencia Tipificación microbiologica Cloned DNA re-sequencing mtDNA Transgenicos Expresion Oligo ID Microsatelite Deteccion de mutaciones Posiciones conocidas Posiciones desconocidas al lado de hot spots Mutaciones puntuales Insercions/deleccions Mutaciones multiples Multiplexing Variacion genetica SNPs y DIPs - Di-, tri- and tetra allelic - Multiple - Multiplexing Haplotipos - Out-of-phase - Allele-specific PCR Cuantificacion Allele frequency assessment - SNP frequency - Tri/tetra allelic SNP frequency - Indel frequency CpG-methylation Gene copy number Loss of heterozygosity Polyploid genomes Expression analyses

109
FIN MUCHAS GRACIAS!

Presentaciones similares










.>")



>")







>")




