Descargar la presentación
La descarga está en progreso. Por favor, espere

2
ENFERMEDADES POR PRIONES
Dr. Alex Espinoza Giacomozzi. Neurología DIPRECA.

3
STANLEY PRUSINER

4
ENCEFALOPATIAS ESPONGIFORMES TRANSMISIBLES
Son enfermedades de humanos y animales que afectan primariamente el SNC No es una respuesta inmune Agentes causales tienen propiedades inusuales

5
Presencia de vacuolización microscópica del tejido cerebral y del depósito de una isoforma anormal, resistente a las proteasas, de una glicoproteína de membrana denominada proteína priónica.

8
Tipos de enfermedades causadas por priones
PORTADOR ENFERMEDAD Humano Enfermedad de Kreutzfeldt- Jakob Sd. De Gerstmann-Strassler-Schenker Kuru Insomnio familiar fatal Ganado Vacuno Encefalopatía espongiforme bovina Oveja, cabra Scrapie Visón Encefalopatía transmisible de visón Mulo, ciervo, alce Enfermedad de debilidad crónica Gatos Encefalopatía espongiforme felina Nyala, kudu mayor Encefalopatía ungulada exótica

10
Patogenia Depósito cerebral de una isoforma patológica (PrPsc), una glucoproteína de membrana codificada por el gen PRNP, situado en el brazo corto del cromosoma 20. Ocurre un cambio de conformación de la isoforma celular normal PrPc . La isoforma anormal tiene un alto contenido en láminas beta y su resistencia parcial a su digestión por proteasas.
, una glucoproteína de membrana codificada por el gen PRNP, situado en el brazo corto del cromosoma 20. Ocurre un cambio de conformación de la isoforma celular normal PrPc . La isoforma anormal tiene un alto contenido en láminas beta y su resistencia parcial a su digestión por proteasas.")
11
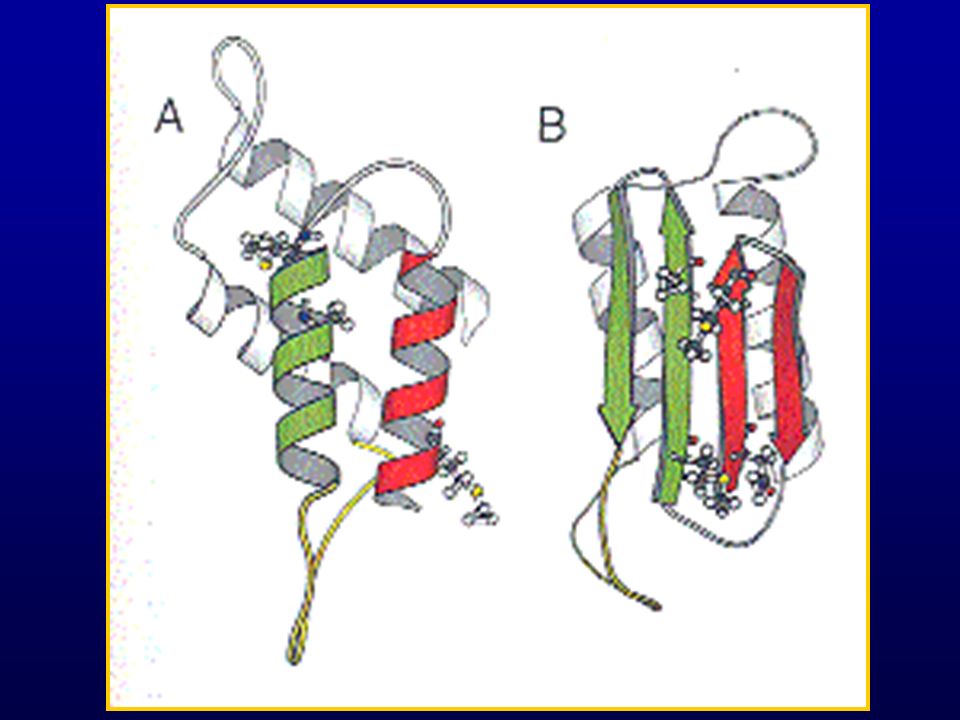
Proteína priónica. Consta de loops helicoidales, dos zonas con hélice beta y una zona de octarepetición. Soluble en detergente no denaturantes. Helicoidal. Sensible a la proteinasa K. 40% más sábanas betas. Insoluble. Resistente a prot K

12
Structure and Function of PrPC
Gene located on Chromosome 20, 250 aa in length, highly -helical Function unknown, suggested: Cell signaling Copper transport, Copper binding

13
PrPC vs PrPSc PrPC PrPSc Sensitive to Proteolysis
Associated with cell membrane (soluble) Mainly helical Resistant to Proteolysis Present in cells as aggregated fibrils (insoluble) Mainly β-sheet Same amino acid sequence, thus conformational difference is not due to amino acid variation.
 Mainly helical. Resistant to Proteolysis. Present in cells as aggregated fibrils. (insoluble) Mainly β-sheet. Same amino acid sequence, thus conformational difference is not due to amino acid variation.")
15
Los agregados se acumulan en el cerebro llevando a la pérdida de neuronas y daño cerebral.
Se acumula en placas. El organismo se defiende en un intento de eliminarlas dejando “hoyos”

18
Pérdida de memoria y agudeza mental, y a Veces compromiso visual Resulta en insomnio Problemas con coordinación Y la marcha Comprometido en bovinos Priones afectan distintas regiones cerebrales, la clínica depende de aquello

19
Encefalopatía espongiforme transmisible subaguda
Creutzfeldt - Jakob Encefalopatía espongiforme transmisible subaguda

20
Tipos: Familiar. Iatrogénica. Esporádica. Variantes “muy” raras.

21
Variantes de ECJ. ECJ atáxica Afectación cerebelosa temprana
Variante de Heidenhain Trastornos visuales tempranos Variante panencefalítica Afectación sust. Gris sust. Blanca Variante amiotrófica Progresiva debilidad muscular inicial.

22
Generalidades Descrita en 1920.
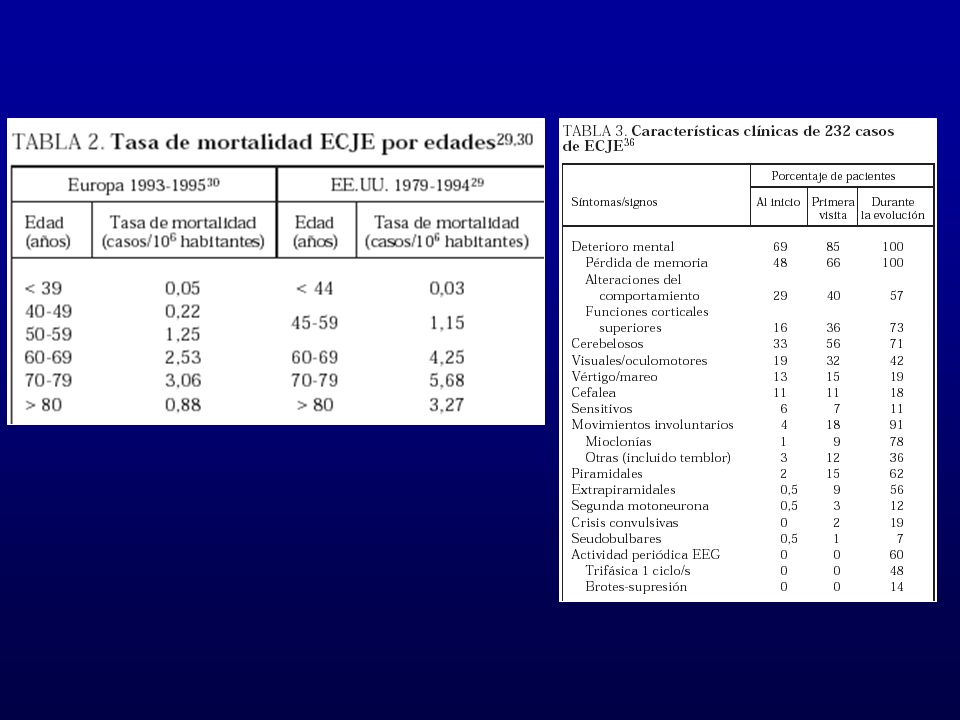
Incidencia 0,5 a 1 casos/ hab. Año No hay distribución estacional. Generalmente 60 –70 años. Mortal: 1 año. Difiere del Kuru y scrapie en epidemiología y patrón clínico.

23
Tipos de ECJ ESPORÁDICA El tipo más común
Aproximadamente el 85% de los casos No hay factores de riesgo Etiología desconocida Conversión espontánea a PrPsc Mutación somática en el gen PRNP Infección exógena

24
ADQUIRIDA/IATROGÉNICA
FAMILIAR 10 a 15 % de los casos Antecedentes familiares de la enfermedad y/o test positivos para la herencia AD. ADQUIRIDA/IATROGÉNICA 0,5 a 1,5 casos por millón Exposición a tejido cerebral, u otro sitio del SN, o fluidos No hay evidencia de contagio por contacto casual

25
Características clínicas
No hay diferencias entre las 3 formas Típico: Demencia rápidamente progresiva Mioclonías Ataxia cerebelosa Presentación clásica: Demencia, mioclonias, EEG ondas periódicas

26
60 % tiene la sintomatología clásica Fase premórbida:
1/3 de los pacientes Fatiga, malestar, trastornos del sueño y alimentarios. Rápido deterioro semana a semana. Duración promedio 6 a 7 meses. 90% mueren en el primer año. Progresión Sd. Demencial y alteraciones conductuales

27
Progresión de síntomas cerebelosos y extrapiramidales.
Rigidez, temblor, coreoatetosis. MIOCLONIAS. Crisis convulsivas y TC Raro: Trastorno oculomotor, parestesias, disfunción vegetativa y neuropatía periférica.

29
ECJ ESPORÁDICA Fase prodrómica: 26 % de los pacientes Inespecífico
CEG y depresión. Astenia, insomnio, anorexia, ansiedad. Psiquiatría. Formas de inicio: 1/3 DOC 1/3 cerebelosa-visual 1/3 mixta Instauración en semanas o meses. 13 a 20% es en días o súbito.

30
DETERIORO MENTAL: Demencia 66% Alteraciones de conducta 40%
Memoria, desorientación espacial, juicio y razonamiento Alteraciones de conducta 40% Agitación, depresión Alteraciones de funciones superiores 36% Anomia, acalculia, agrafia

31
Fase de estado: Estadio final 91% movimientos anormales
Mioclonías 78% espontáneas y reflejas 62% piramidalismo 56% extrapiramidales Estadio final Deterioro global de funciones superiores. mutismo

32
Duración de la enfermedad:
Casos descritos desde 3 semanas a 10 años. Media 8 meses 70% fallece antes de 6 meses 90% antes del<año MORTAL

34
ECJ FAMILIAR CHILE 10 a 15 % de casos Herencia autosómica dominante
Edad inicio más precoz, curso más lento EEG típico no siempre se encuentra Proteína (-) 50% Mutación más común es en el codón 200 Judios Lybian en Israel CHILE
 50% Mutación más común es en el codón 200. Judios Lybian en Israel. CHILE.")
35
ECJ IATROGENICA 1974 mujer que recibió transplante corneal
Donante falleció de ECJ Uso de duramadre cadavérica Electrodos de EEG estereotáxico Hormona pituitaria

36
NUEVA VARIANTE ECJ Entre 1994 y 1997 22 casos reportados.
Inicio más joven (29 años). Signos psiquiatricos precoces, disestesias , parestesias. Curso prolongado. EEG no típico
. Signos psiquiatricos precoces, disestesias , parestesias. Curso prolongado. EEG no típico.")
37
Placas similares a Kuru.
Ninguno tenia mutación del PrP, pero todos eran homocigotos para metionina en codon 129. Todos comían carne, excepto 1. Enfermedad bovina y nv ECJ comparten características en el tejido cerebral

38
DIAGNÓSTICO LCR: Elevación moderada de proteínas < 100 mg/dl
1996 Hsich en NEJM: proteína No es específica de las encefalopatías espongiformes transmisibles Puede preceder a actividad EEG y detectarse en estadios precoces S: 94% E: 84% VVP:97% VVN: 72%

40
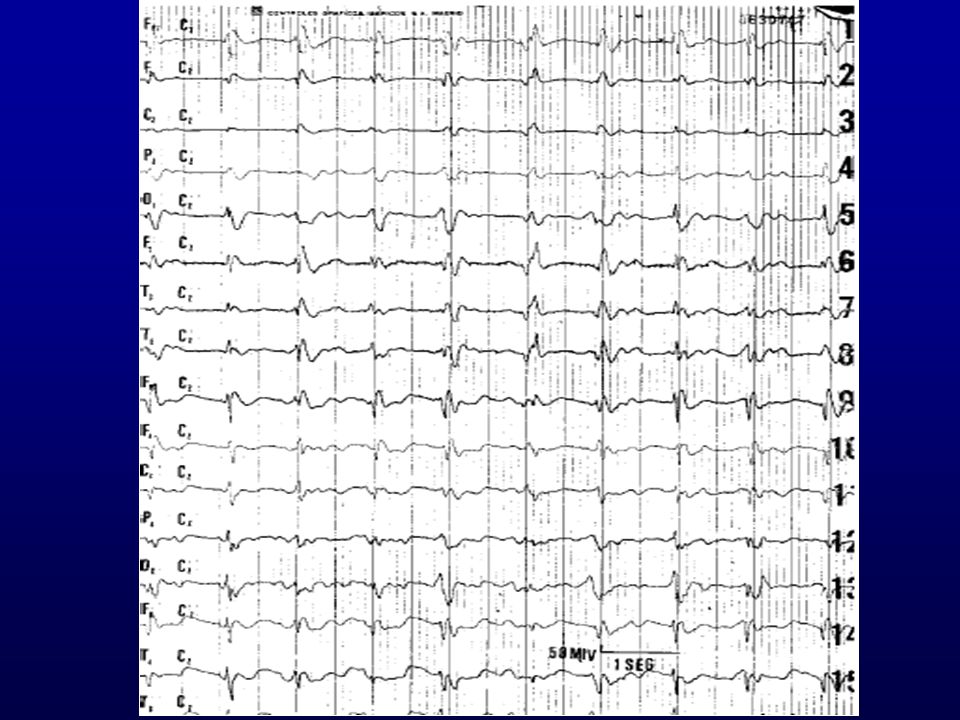
ELECTROENCEFALOGRAMA:
Típico: Actividad periódica generalizada de ondas agudas bi o trifásicas de 100 a 600 ms de duración que se repiten a un intervalo de ms sobre un trazado de fondo enlentecido con actividad delta o theta. Depende de momento de la enfermedad y nº de EEG S: 66% E: 74% Semanal

42
RESONANCIA MAGNETICA:
Hiperseñal en caudado y putamen , bilateral y simetrica en T2 y densidad protonica S:67% E:93% Atrofia cortical en el 29% BIOPSIA Gold standard

43
MANEJO No existe tratamiento especifico efectivo para curarla ni controlarla. Se han probado amantadina, aciclovir, interferon, esteroides, Etc. Manejo general.

44
RIESGO DE TRANSMISION Precauciones universales
No se transmite por contacto directo No se transmite por via aerea No hay paso transplacentario LCR: doble guante, proteccion ocular. Extremadamente dificil de eliminar Esterilización normal no lo elimina

45
Instrumentos deben ser descontaminados por lavado en 1 N hidroxido de sodio (40 gr por litro) o hipoclorito de sodio no diluido por una hora y luego en autoclave a 134 grados Celsius por una hora Fijados y lavados en acido formico concentrado por 1 hora y luego en formaldehido al 4% por 48 horas.

46
Medida generales Atención a pacientes
Limpieza, descontaminación y esterilización de material no desechable. Medidas específicas en función del servicio o procedimiento empleado Servicios Médicos Neurología y Electrofisiología Electrodos cutáneos para EEG, soportes plásticos de electrodos. Esterilizados o eliminados. Electrodos SC o IM para EMG o intracraneales deben ser destruidos.

47
Sd. Gerstmann-Strassler-Schenker
Descrito en 1936 Trastorno familiar. Hay casos esporadicos 2 por cada Ataxia de tronco y extremidades, demencia, y a veces, parkinsonismo. Duracion: 1 a 11 años. Media 5 años Deposito placa amiloide, degeneracion sustancia blanca, cambios espongiformes, gliosis Corteza cerebelosa y corteza cerebral.

48
Insomnio familiar fatal
Autosómico dominante. Sueño progresivo y trastornos autonómicos. Alucinaciones complejas hasta estupor y coma Taquicardia, hiperhidrosis e hiperpirexia Trastornos circadianos Ataxias, mioclonias y otros sts motores. Perdida neuronal y gliosis del talamo, sustancia gris cortical, centro oval, cerebelo y oliva inferior.

49
Kuru 1955:Un explorador australiano en Nueva Guinea describió a una niña temblando y riendo. 1956: Carelton Gadusek fue a Nueva Guinea a estudiar el “Kuru”: temblor y sacudidas. Etnia Fore en Papua Nueva Guinea. Kuru en idioma fore: temblores de frío y miedo.

50
Ritual religioso. Al educar y eliminar el ritual disminuyó la incidencia Generaciones de 1959 no se conoce ningún nuevo caso Latencia prolongada hasta 3 décadas Edad de comienzo 5 a 60 años Ataxia cerebelosa, demencia y signos extrapiramidales.

51
Bibliografía La enfermedad de Creutzfeldt-Jakob esporádica: variabilidad fenotípica M.J. Moreno y J. Romero Neurología 2002;17(7):366-77 Historia y clasificación de las enfermedades priónicas humanas J.M. Polo REV NEUROL 2000; 31 (2): Revisión de la Enfermedad de Creutzfeldt Jakob u otras enfermedades priónicas. Revneurol (12):
: Revisión de la Enfermedad de Creutzfeldt Jakob u otras enfermedades priónicas. Revneurol (12):")
Presentaciones similares























>")




