Descargar la presentación
La descarga está en progreso. Por favor, espere

1
Dra. Judith Izquierdo Medicina Interna

2
La enfermedad de Wilson (EW), también denominada degeneración hepatolenticular, es un transtorno hereditario caracterizado por alteraciones en el metabolismo del cobre por un déficit en el aclaramiento biliar, y secundariamente su acumulación en órganos diana.
, también denominada degeneración hepatolenticular, es un transtorno hereditario caracterizado por alteraciones en el metabolismo del cobre por un déficit en el aclaramiento biliar, y secundariamente su acumulación en órganos diana.")
3
Su incidencia se estima en 1 caso por 30.000 nacidos vivos y la prevalencia de la enfermedad se estima en 10 a 30 casos por millón de habitantes.

4
La EW es de herencia autosómica recesiva El cromosoma 13 (alelo q14.3-q21) el que contiene el gen ATP7B que codifica una proteína ATPasa responsable de la excreción biliar de cobre y la unión del cobre a la ceruloplasmina, transportadora de cobre, con expresión de esta ATPasa en múltiples tejidos La ATP7B es una proteína transmembrana con 8 segmentos y 6 zonas de unión al cobre Se han descrito hasta 200 mutaciones del gen
 el que contiene el gen ATP7B que codifica una proteína ATPasa responsable de la excreción biliar de cobre y la unión del cobre a la ceruloplasmina, transportadora de cobre, con expresión de esta ATPasa en múltiples tejidos La ATP7B es una proteína transmembrana con 8 segmentos y 6 zonas de unión al cobre Se han descrito hasta 200 mutaciones del gen")
5
El cobre es un oligoelemento esencial para el organismo, ya que participa de acciones de la vida celular, como generar energía mitocondrial, regular el transporte tisular del hierro, formar melanina, eliminar radicales libres de oxígeno y madurar el tejido conectivo a través de sus acciones como coenzima

8
Neurológicas Hepáticas Edad de presentación desde los 10 a los 50 años

9
Elevación de transaminasas Hepatopatía crónica ( Cu: induce la fibrogénesis, infiltra los hepatocitos e induce la inflamación portal, conduciendo a la cirrosis) Cirrosis hepática Fallo hepático fulminante (mujeres, liberación masiva Cu al torrente sanguíneo…. Hemolisis IV) Infrecuente hepatocarcinoma
 Infrecuente hepatocarcinoma.")
11
Presentes en el 30% casos Aparecen en la segunda a tercera década de la vida Sintomas y signos extrapiramidales y bulbares (temblor, rigidez, coreoatetosis, rigidez, disartria o disfagia) conservando las funciones superiores.
 conservando las funciones superiores.")
12
Aparece por el depósito de cobre en la membrana de Descemet, inicialmente en los polos superiores e inferiores de la córnea y con posterioridad rodeando el iris

13
Manifestada en el 20% Presencia de alteraciones del comportamiento, disminución del rendimiento y manifestaciones psicóticas o neuróticas, pudiendo ser causa también de demencia orgánica.

14
renales: síndrome de Fanconi o acidosis tubular renal hematológicas: anemia hemolítica no autoinmune, leucopenia y trombopenia óseas: suele observarse osteopenia y osteoporosis, artropatía con formación de quistes subcondrales y condrocalcinosis, así como roturas musculotendinosas y calcificacione heterotópicas otras: miocardiopatías y arritmias fatales, cataratas sunflower por depósito del metal en el cristalino, rabdomiólisis, acantosis nigricans, intolerancia a los hidratos de carbono, amenorrea.

17
HC. E.F. Cobre y ceruloplasmina Los niveles disminuidos de ceruloplasmina en plasma (< 20 mg/dl) suelen considerarse diagnósticos en caso de que estén acompañados de anillo de Kaiser-Fleischer, *una proporción de pacientes presentan niveles de ceruloplasmina normales o discretamente disminuidos Pruebas de función hepática. Elevación de transaminasas
 suelen considerarse diagnósticos en caso de que estén acompañados de anillo de Kaiser-Fleischer, *una proporción de pacientes presentan niveles de ceruloplasmina normales o discretamente disminuidos Pruebas de función hepática. Elevación de transaminasas.")
19
Resonancia magnética cerebral Permite identificar depósitos de cobre a nivel del tálamo y ganglios basales, así como el grado de atrofia cortical.

20
Biopsia hepática Nos permite identificar el grado de hepatopatía que se padece, esteatosis o fibrosis. La determinación de cobre hepático en las muestras de biopsia es útil incluso en pacientes que no han desarrollado sintomatología alguna, observándose niveles elevados por encima de 250 /g de su peso seco (normal < 50 g/g)
.")
21
Test genético El análisis con técnicas de reacción en cadena de la polimerasa (PCR) de ADN se han de solicitar siempre que exista sospecha alta o los estudios avalen esta enfermedad. También es importante el cribado de familiares de primer grado menores de 40 años.

22
Medidas higiénico-dietéticas Evitar agua y alimentos ricos en cobre como son vísceras de animales, chocolate, cacao, frutos secos, marisco, soja, gelatinas y setas. Nunca será una medida aislada.

23
D-penicilamina La unión del cobre a la penicilamina forma complejos que son eliminados por la orina, actuando por tanto de quelante del cobre. Es necesario añadir piridoxina (vitamina B6) diaria, ya que este fármaco puede inducir su deficiencia. Efectos adversos: hipersensibilidad, lupus like, proteinuria en rango nefrótico, síndrome de Goodpasture o cuadros de toxicidad medular como anemia Hasta en el 50% de los casos, instaurado el tratamiento se produce un empeoramiento de la clínica neurológica, lo que obligará a disminuir la dosis y al incremento secuencial.
 diaria, ya que este fármaco puede inducir su deficiencia. Efectos adversos: hipersensibilidad, lupus like, proteinuria en rango nefrótico, síndrome de Goodpasture o cuadros de toxicidad medular como anemia Hasta en el 50% de los casos, instaurado el tratamiento se produce un empeoramiento de la clínica neurológica, lo que obligará a disminuir la dosis y al incremento secuencial..")
24
Trientine Su mecanismo es doble, al favorecer la eliminación del cobre por la orina y bloquear su absorción a nivel intestinal Menos efectos secundarios neurológicos que la d- penicilamina Zinc Las sales de zinc aumentan los niveles de metalotioneínas, proteínas con gran afinidad por el cobre, aumentando su eliminación por heces. Empleo durante las formas asintomáticas de la enfermedad y el embarazo. Presenta buena tolerancia.

25
Tetratiomolibdato de amonio Bloquea la absorción intestinal del cobre dietético y favorece la unión del cobre a la albúmina, bloqueando la entrada al interior de la célula y evitando por tanto su toxicidad. Trasplante hepático Su indicación es en pacientes que han desarrollado insuficiencia hepática fulminante o cirrosis en estadio final con las complicaciones derivadas de la hipertensión portal y sin respuesta al tratamiento quelante (controvertido en pacientes con clínica neurológica aislada). No hay evidencia de recidiva tras el trasplante.
. No hay evidencia de recidiva tras el trasplante..")
27
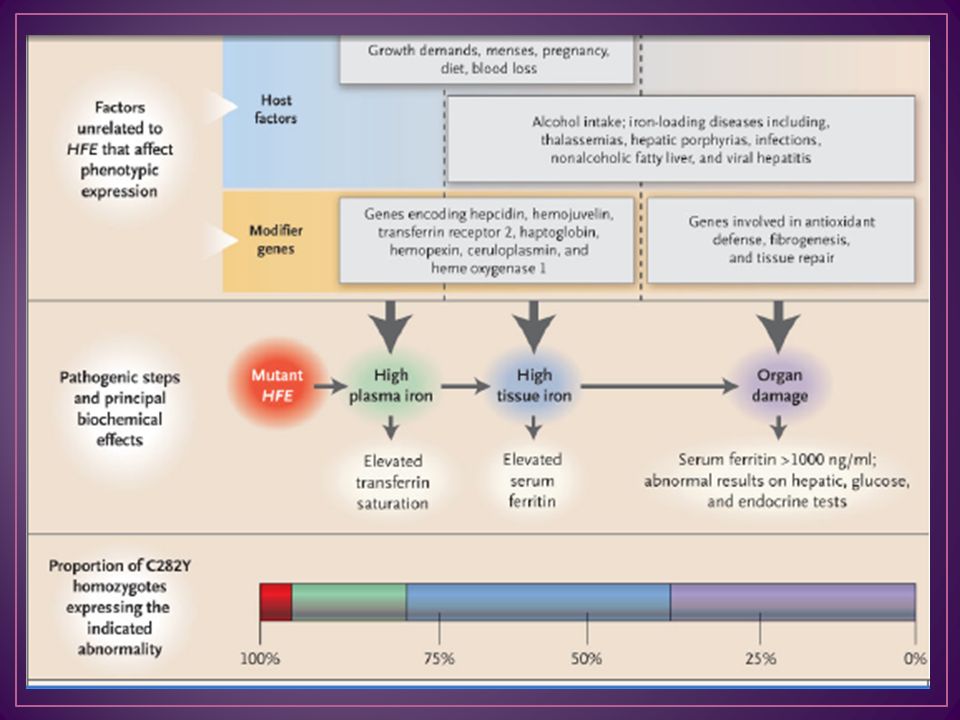
Enfermedad autosómica recesiva en la que un incremento en la absorción de hierro causa el depósito progresivo de este metal, que puede resultar en daño tisular y que puede llegar a ser fatal

28
Mutación C282Y (sustitución de cisteína por tirosina) del gen HFE situado en el brazo corto del cromosoma 6, 80% de pacientes H63D (histidina por aspartato) Homocigota
 del gen HFE situado en el brazo corto del cromosoma 6, 80% de pacientes H63D (histidina por aspartato) Homocigota")
30
Fe se mantiene entre 3 y 4 g En la HC la absorción supera 4 o más veces lo normal, pudiendo los depósitos de Fe alcanzar 20 g aumento de la velocidad de transporte del metal desde el enterocito hacia la circulación disminución de la capacidad de almacenamiento de Fe en el sistema reticuloendotelial, con lo que el metal sería transferido predominantemente a los parénquimas Fe unido a la transferrina, en lugar de ser liberado preferentemente a la médula ósea, lo hace al parénquima hepático

35
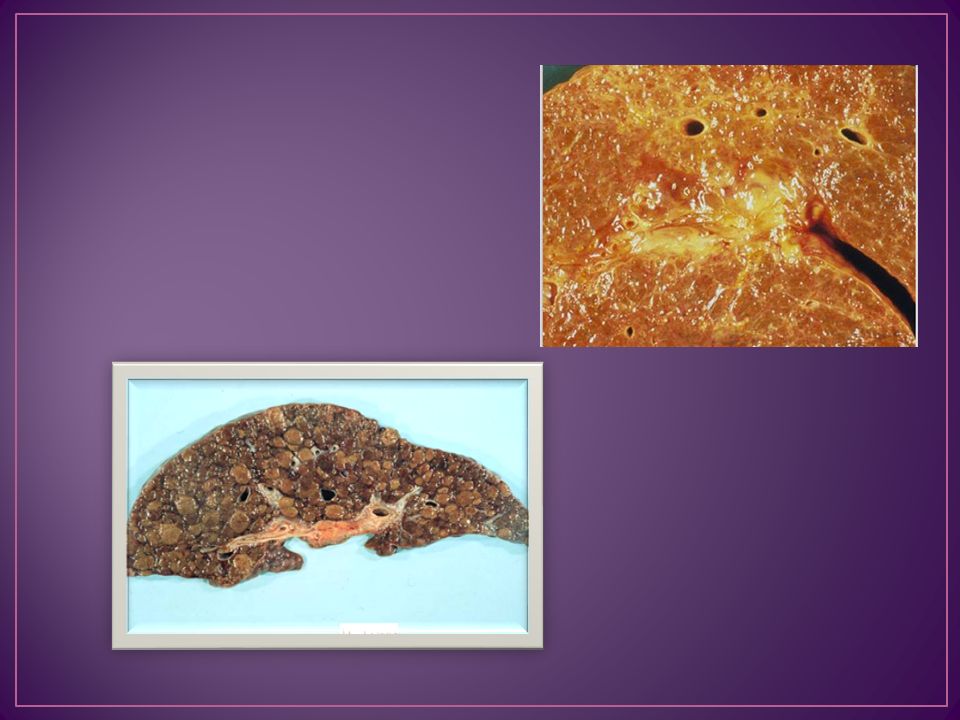
aumento progresivo de gránulos de hemosiderina en los hepatocitos, preferentemente periportales páncreas exocrino y endocrino Tiroides Suprarrenales Adenohipófisis En testículo existe atrofia del epitelio germinal y ausencia de células de Leydig

36
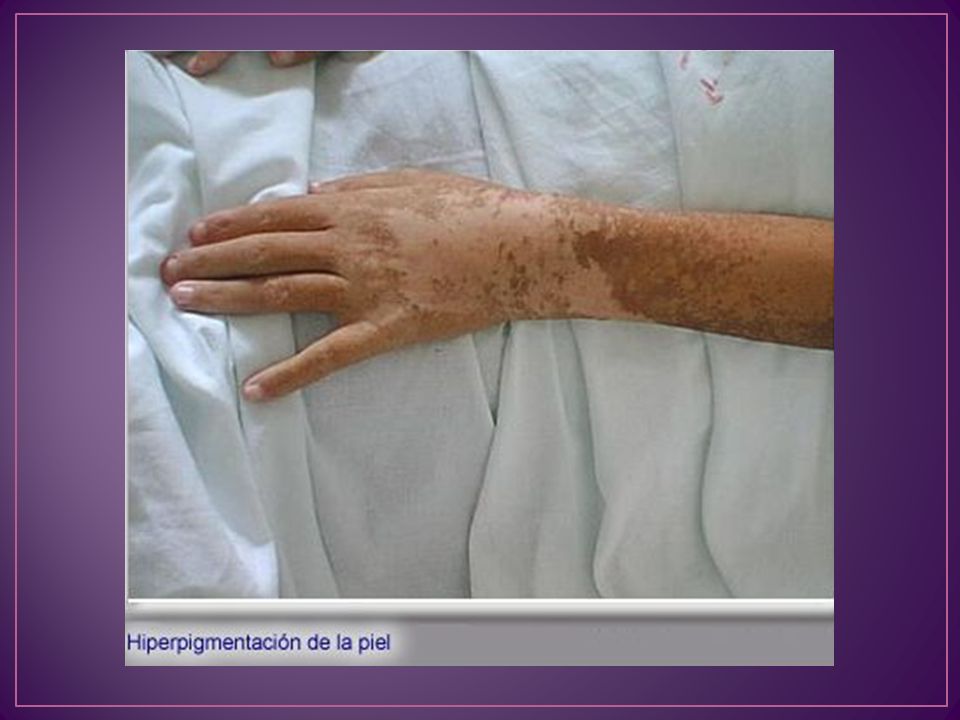
En el miocardio se aprecia Fe en el citoplasma de las fibras contráctiles y del sistema de conducción. También se detecta hemosiderina en las células sinoviales con fibrosis La piel muestra un incremento de melanina en la epidermis basal, y de hemosiderina en los macrófagos y glándulas sudoríparas

37
50 años hepatomegalia aparece en el 90% de los casos con cirrosis establecida 80% se detecta intolerancia a los hidratos de carbono miocardiopatía con arritmias e insuficiencia cardíaca Las artralgias como síntoma inicial aparecen aproximadamente en el 50% La hiperpigmentación sucede en el 75%

39
regla de las tres A: astenia crónica Artralgias transaminasas (aminotransferasas) incremento inferior a 3 veces lo normal
 incremento inferior a 3 veces lo normal")
40
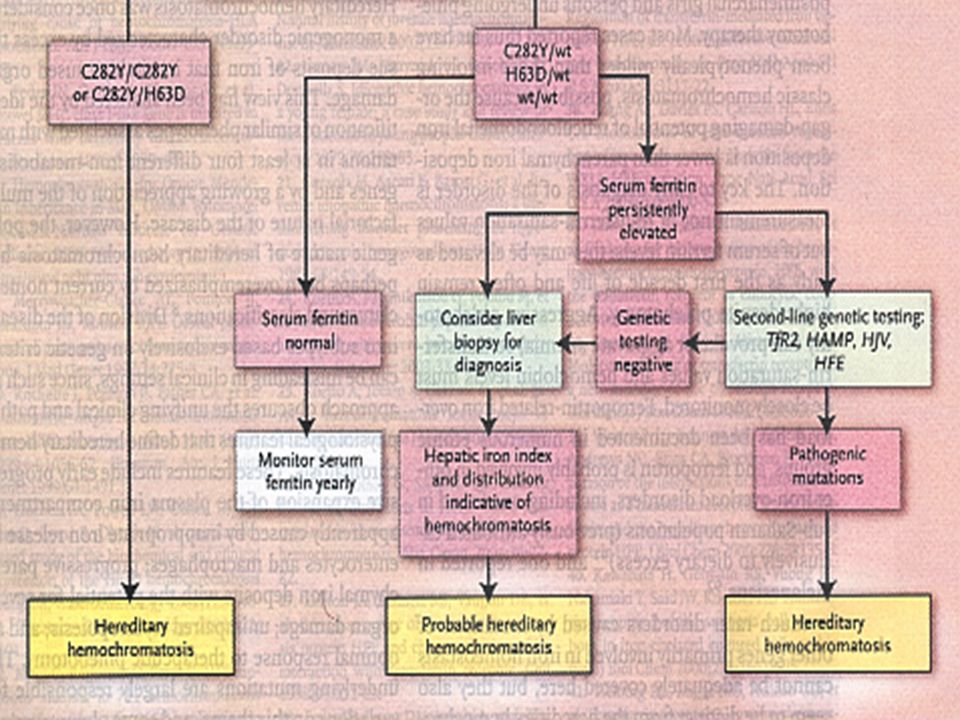
estudio del metabolismo del Fe saturación de la transferrina superior al 60% en el varón y al 50% en la mujer ferritina sérica están habitualmente elevados 1.000 y 6.000 ng/mL La confirmación del diagnóstico se basa en la biopsia hepática con tinción de Perls y cuantificación de Fe en mg/g de tejido seco

41
detección de la mutación C282Y del gen HFE

44
Flebotomías Se extraen 500 mL una o dos veces a la semana (cada una equivale a 250 mg de Fe), durante 2 a 3 años en los casos con intensa sobrecarga férrica
, durante 2 a 3 años en los casos con intensa sobrecarga férrica")
45
Desferroxiamina subcutánea o intravenosa (10-20 mg/día). la diabetes la cardiopatía insuficiencia gonadal cirrosis

Presentaciones similares
























. Forma primaria: herencia autosómica recesiva Alteración.>")
 y electrones (citocromos)>")





