Descargar la presentación
La descarga está en progreso. Por favor, espere

1
Síntesis y degradación
Bioquímica hepática El metabolismo del glucógeno. Síntesis y degradación

2
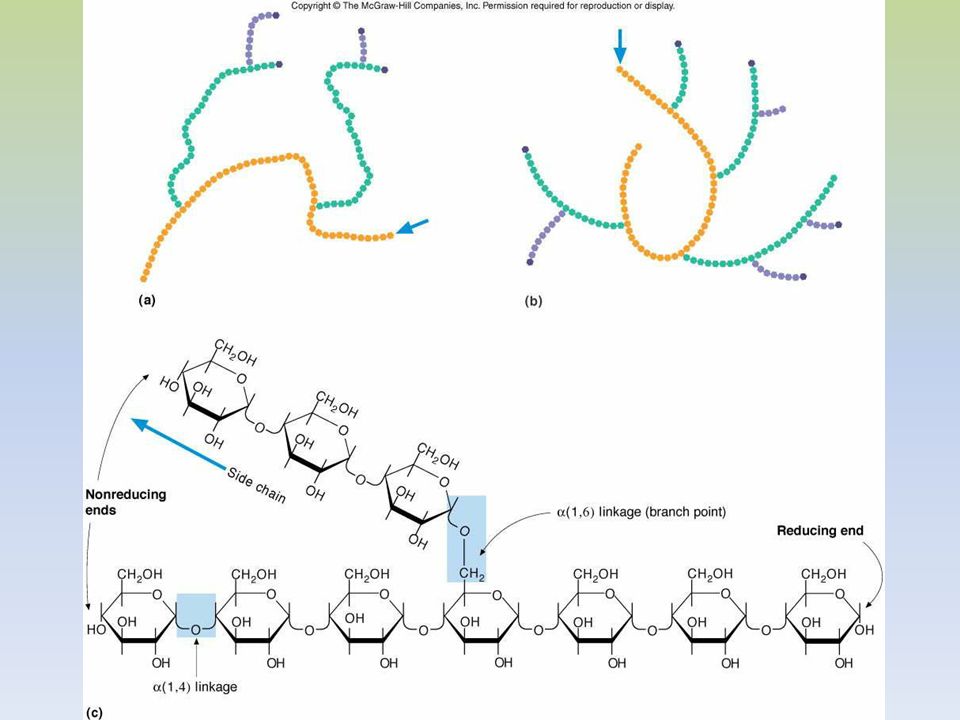
EL GLUCÓGENO ES UN POLÍMERO MACROMOLECULAR, ALTAMENTE RAMIFICADO.
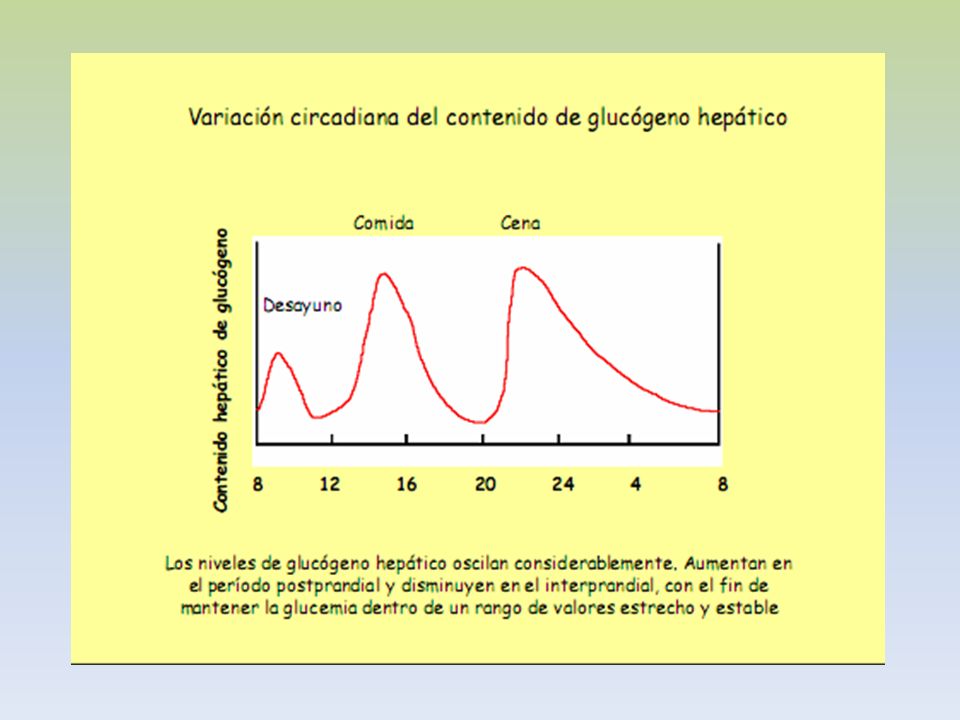
SOLO GLUCOSA ES SU CONSTITUYENTE BÁSICO. PRESENTA UNIONES ALFA 1,4 Y ALFA 1,6. EXISTE GLUCÓGENO HEPÁTICO Y MUSCULAR. EN HÍGADO EL GLUCÓGENO SE ENCUENTRA EN EL CITOPLASMA, EN FORMA DE GRÁNULOS, EN LOS QUE TAMBIEN SE ENCUENTRAN LAS ENZIMAS DE SU METABOLISMO. LA CANTIDAD DE GLUCÓGENO HEPÁTICO, CONTRIBUYE A REGULAR EL NIVEL DE GLUCOSA EN LA SANGRE, YA QUE PROVEE UN DEPÓSITO DE GLUCOSA QUE ES DE FÁCIL ACCESO, EN CASO DE NECESIDAD.

12
-La insulina que se libera del páncreas en respuesta a una elevación de la glucemia, desencadena una cascada de activación de la fosfoproteína fosfatasa. -Esta fosfatasa cataliza la hidrólisis de los grupos fosfato de todos los enzimas implicados en el metabolismo del glucógeno (fosforilasa, fosforilasa quinasa, sintetasa). -El resultado es la inactivación de la glucogenolisis y la activación de la glucogenogénesis. -La síntesis eficaz de glucógeno tendrá lugar en la medida haya UDP-glucosa disponible. -La insulina antagoniza los efectos del glucagón, porque aumenta el transporte de glucosa hacia los tejidos.
. -El resultado es la inactivación de la glucogenolisis y la activación de la glucogenogénesis. -La síntesis eficaz de glucógeno tendrá lugar en la medida haya UDP-glucosa disponible. -La insulina antagoniza los efectos del glucagón, porque aumenta el transporte de glucosa hacia los tejidos.")
14
GLUCOGENOSIS TRASTORNOS EN EL METABOLISMO DEL GLUCÓGENO POR AUSENCIA DE ENZIMAS QUE PARTICIPAN EN SU BIOSÍNTESIS Y DEGRADACIÓN SE TRADUCE EN UNA ANOMALÍA CUANTITIATIVA O CUALITATIVA DEL GLUCÓGENO DEPOSITADO. SEGÚN EL ÓRGANO AFECTADO LAS GLUCOGENSOSIS SE CLASIFICAN EN: I) HEPÁTICA O HEPATORRENAL: I, III, IV, VI, X HEPATOMEGALIA, HIPOGLUCEMIIA, HIPERTG, HIPERCOLESTEROLEMIA, LACTATO Y ÁCIDO ÚRICO. MUERTE INFANCIA II) CARDÍACA O GENERALIZADA: II INFILTRACIÓN MASIVA CON GLUCÓGENO, ESPECIALMENTE MIOCARDIO, FUNCIÓN MUSCULAR MUERTE FALLA CARDÍACA III) MIOPÁTICA: V, VII, VIII, IX - CALAMBRES, CANSANCIO, ATROFIA MUSCULAR
 HEPÁTICA O HEPATORRENAL: I, III, IV, VI, X. HEPATOMEGALIA, HIPOGLUCEMIIA, HIPERTG, HIPERCOLESTEROLEMIA, LACTATO Y ÁCIDO ÚRICO. MUERTE INFANCIA. II) CARDÍACA O GENERALIZADA: II. INFILTRACIÓN MASIVA CON GLUCÓGENO, ESPECIALMENTE MIOCARDIO, FUNCIÓN MUSCULAR MUERTE FALLA CARDÍACA. III) MIOPÁTICA: V, VII, VIII, IX. - CALAMBRES, CANSANCIO, ATROFIA MUSCULAR.")
16
GLUCOGENOSIS TIPO I: ENFERMEDAD DE VON GIERKE
•Incapacidad de liberar glucosa al torrente sanguíneo a partir de Glucosa-6-P •Déficit de G-6-Pasa - AR •Manifestación hepática: hepatomegalia, riñón agrandado •Hipoglucemia severa durante el ayuno inhibe la liberación de insulina •Aumento de la glucolisis hepática, sin liberación de glu al torrente sanguíneo •Acidemia severa hipoglucemia estimula la liberación de catecolaminas y facilita la degradación de glucógeno muscular a ácido láctico. • Infiltración grasa por liberación de lípidos como fuente de energía Menor síntesis de proteínas crecimiento retardado. Grave

18
TRATAMIENTO: - INGESTIÓN FRECUENTE DE ALIMENTOS Y UN RÉGIMEN RICO EN PROTEÍNAS EVITARÍA O DISMINUIRÍA EL DEPÓSITO DE GLUCÓGENO. ACTH O CORTISONA PARA REGULAR LOS EPISODIOS DE HIPOGLUCEMIA. PARA EVITAR LA ACIDOSIS ADM LACTATO DE SODIO. DIAGNÓSTICO: SE DEMUESTRA LA AUSENCIA DE LA ENZIMA EN TEJIDO HEPÁTICO POR BIOPSIA O EXÁMEN POST-MORTEM. PRUEBAS DE TOLERANCIA: A LA GALACTOSA, FRUCTOSA, GLUCAGÓN Y EPINEFRINA NO SE OBSERVA AUMENTO DE LA GLUCEMIA

19
GLUCOGENOSIS TIPO II: ENFERMEDAD DE POMPE
•Ausencia de alfa 1-4 glucosidasa lisosomal – AR Se manifiesta en la primera infancia Síntomas clínicos: vómitos, anorexia, retardo de crecimiento, debilidad muscular, cianosis, disnea. Las alteraciones metabólicas no son muy pronunciadas dado que las otras vías metabólicas del glucógeno no están afectadas •Cardiomegalia que puede producir la muerte a edad temprana por fallo cardíaco Observaciones físicas: apariencia de imbecilidad, hipotonía muscular, hipertrofia cardíaca, alteraciones neurológicas, no se observa hepato-esplenomegalia. •Acumulación de rosetas de glucógeno no degradadas en el interior de los lisosomas • No hay tratamiento

20
Diagnóstico: La glucemia y las pruebas de tolerancia dan todas normales Cuadro hematológico: normal, la tinción de leucocitos para identificar glucógeno revela acumulación del mismo. Alteraciones cardiográficas No se observa: hipoglucemia, acidosis y cetosis, lípidos plasmáticos normales. Se observa acumulación de glucógeno en corazón y músculo.

23
GLUCOGENOSISTIPO III:
•Ausencia del enzima desramificante amil-1,6-glucosidasa - AR •El glucógeno solo se puede degradar parcialmente por la glucógeno fosforilasa, no más allá de la dextrina límite. •Manifestaciones clínicas: Afectados hígado y músculo Más moderadas que en la enfermedad de von Gierke. Hipoglucemia menos severa •Acumulación de glucógeno hepático, que tiene cadenas externas cortas. TRATAMIENTO: - Dieta rica en proteínas la transaminación y conversión de aminoácidos en intermediarios glucolíticos puede corregir el déficit primario.

24
DIAGNÓSTICO: Si damos Gal o Fru aumenta la glucemia porque está la Glu-6-Pasa y puede pasar a Glu ( a la tipo I) DOBLE PRUEBA DEL GLUCAGÓN: La administración de glucagón no aumenta la glucemia en la tipo I. En la tipo III A) La prueba se hace luego de una hora de ayuno glucógeno se encuentra ramificado si es tipo III aumenta la glucemia B) después de 14 hs de ayuno, se reanuda la adm de glucagón no aumenta la glucemia porque por la primera prueba, el glucógeno llegó a los puntos de ramificación. DETERMINACION ENZIMÁTICA: SUSTRATO ARTIFICIAL PENTASACÁRIDO (4 GLU- ALFA 1,4 Y UNA GLU TERMINAL CON UNIÓN ALFA 1,6
 La prueba se hace luego de una hora de ayuno glucógeno se encuentra ramificado si es tipo III aumenta la glucemia. B) después de 14 hs de ayuno, se reanuda la adm de glucagón no aumenta la glucemia porque por la primera prueba, el glucógeno llegó a los puntos de ramificación. DETERMINACION ENZIMÁTICA: SUSTRATO ARTIFICIAL PENTASACÁRIDO (4 GLU- ALFA 1,4 Y UNA GLU TERMINAL CON UNIÓN ALFA 1,6.")
25
GLUCOGENOSIS TIPO IV: DEFICIT DE LA ENZIMA RAMIFICANTE O AMILOPECTINOSIS NO SE SABE SI ES AR O LIG A X AUSENTE LA ENZIMA AMILO 1,4- 1,6 – TRANSGLICOSIDASA SÍNTOMAS CLINICOS: AL AÑO DE EDAD CIRROSIS HEPÁTICA, HÍGADO GRANDE, NODULAR, ESPLENOMEGALIA. PRUEBAS FUNCIONALES HEPÁTICAS ANORMALES CARACTERÍSTICAS ESTRUCTURALES DEL GLUCÓGENO AISLADO DE TEJIDOS: Similar a la amilopectina, es decir con menor número de puntos ramificados (comparado con el glucógeno normal) Menos soluble que el glucógeno normal Cirrosis por reacción contra un cuerpo extraño glucógeno precipitado. No se observa acumulación de glucógeno en tejidos y GR. Grave el paciente muere antes de los 5 años. TRATAMIENTO: SINTOMÁTICO Y PALIATIVO
 Menos soluble que el glucógeno normal. Cirrosis por reacción contra un cuerpo extraño glucógeno precipitado. No se observa acumulación de glucógeno en tejidos y GR. Grave el paciente muere antes de los 5 años. TRATAMIENTO: SINTOMÁTICO Y PALIATIVO.")
26
DIAGNÓSTICO: - Si se mide la absorción de amilopectina con I2 se observa un pico máximo a 550 nm El glucógeno de los enfermos + I2 presenta un pico a 530 nm. El glucógeno normal + I2 nm la estructura del glucógeno del enfermo es similar a la amilopectina La enzima tb se encuentra ausente en leucocitos se hacen ensayos “in vitro”, para demostrar la ausencia de esta enzima Las respuestas a la adrenalina, glucagón y sobrecarga a la glu son anormales.

28
GLUCOGENOSISTIPO V: ENFERMEDAD DE MC ARDLE
• AR - Ausencia de fosforilasa muscular Fosforilasa hepática normal distinto control genético •No se moviliza el glucógeno muscular como consecuencia del ejercicio, su estructura es normal. En esta enfermedad faltan las fosforilasas a (activa) y b (inactiva) SÍNTOMAS CLÍNICOS: Incapacidad para el esfuerzo físico, astenia, disfagia. Los paciente sufren calambres y la imposibilidad de realizar ejercicios mínimamente enérgicos •No se observa el aumento de lactato sérico típico después de un esfuerzo muscular •Daño muscular (distrofia) como consecuencia de un metabolismo energético inadecuado •Aumento de marcadores de lesión muscular (CPK, aldolasa, mioglobina). TRATAMIENTO: No hay
 y b (inactiva) SÍNTOMAS CLÍNICOS: Incapacidad para el esfuerzo físico, astenia, disfagia. Los paciente sufren calambres y la imposibilidad de realizar ejercicios mínimamente enérgicos. •No se observa el aumento de lactato sérico típico después de un esfuerzo muscular. •Daño muscular (distrofia) como consecuencia de un metabolismo energético inadecuado. •Aumento de marcadores de lesión muscular (CPK, aldolasa, mioglobina). TRATAMIENTO: No hay.")
29
DIAGNÓSTICO: Un enfermo no puede hacer flexiones profundas de rodillas durante más de 1 minuto. Se hace la biopsia muscular y se mide la concentración de glucógeno y actividad de fosforilasa. PRUEBA DEL EJERCICIO ANÓXICO: Se insufla el manguito del aparato de presión arterial por encima de la presión sistólica. A- Se obtiene una muestra de sangre venosa antes del ejercicio. B- Se abre y se cierra el puño durante un minuto. C- Se extrae nuevamente sangre D- Se repite 3 veces Los niveles sanguíneos de lactato se elevarán aproximadamente el 3 veces en un sujeto normal En el paciente no se observa lo mismo queda su mano en posición tetánica

31
GlucogenosisTipo VI: •Ausencia de fosforilasa hepática – AR •No se moviliza el glucógeno hepático •La hipoglucemia moderada en ayunas, compensada en parte por gluconeogénesis Lípidos moderadamente aumentados Glucógeno en GR aumentado Äcido úrico aumentado La administración de Gal, aumenta la glucemia DIAGNÓSTICO: Deter la actividad de fosforilasa en leucitos e hígado Cuantificar contenido de glucógeno. La activ de la enzima es normal luego de la adm de glucagón TRATAMIENTO: - Dieta rica en proteínas y adm de zinc- glucagón La fosforilasa hepática es genéticamente diferente a la muscular.

32
GLUCOGENOSIS TIPO VII:
Deficiencia de la fosfofructoquinasa muscular Dolores musculares DIAGNÓSTICO: El músculo contiene mas del doble de glucógeno que lo normal. Su estructura es normal La producción de lactato a partir de glucógeno, Glu- 1-P y Fru-1-P es baja. Su producción desde Fru-1,6 diP es normal La ausencia de la enzima ha sido determinada por reacción inmunológica. GLUCOGENOSIS TIPO VIII: Deficiencia de fosfoglucomutasa Hay acumulación de glucógeno en músculo Hepatomegalia GLUCOGENOSIS IX: Deficiencia de fosfohexosa isomerasa muscular Se mide lactato con distintos sustratos usando homogenatos de músculo.

33
GLUCOGENOSIS TIPO X: Deficiencia de fosforilasa quinasa de hígado AGLUCOGENOSIS: Ausencia de glucógeno sintetasa Bajos niveles de glucógeno Hipoglucemia, marcada en ayunas Deficiencia mental secundaria a la hipoglucemia Alteración en el metabolismo de los lípiods Diagnóstico: biopsia ausencia de glucógeno y glucógeno sintetasa

Presentaciones similares