Descargar la presentación
La descarga está en progreso. Por favor, espere

1
NEOPLASIAS LINFOIDES Prof. Germán Detarsio Cátedra de Hematología
Facultad Cs. Bioquímicas y Farm. UNR

2
NEOPLASIAS LINFOIDES Son procesos de naturaleza clonal, surgidos de una mutación de la stem cell comprometida para la progenie linfoide (B ó T). Fenómeno observable tanto en sangre periférica como en MO.

3
CLASIFICACION DE LAS NEOPLASIAS LINFOIDES
Células B Células T

4
NEOPLASIAS DE CÉLULAS B MADURAS
LLC B / LINFOMA A CELULAS PEQUEÑAS LEUCEMIA PROLINFOCÍTICA B LEUCEMIA A CELULAS VELLOSAS Ó HAIRY CELL LEUKEMIA (HCL) / LINFOMA A CELULAS VELLOSAS MIELOMA / PLASMOCITOMA MACROGLOBULINEMIA DE WALDESTRÖM
 / LINFOMA A CELULAS VELLOSAS. MIELOMA / PLASMOCITOMA. MACROGLOBULINEMIA DE WALDESTRÖM.")
5
NEOPLASIAS DE CÉLULAS T MADURAS
LEUCEMIA PROLINFOCITICA T (LPLT) LEUCEMIA A LINFOCITOS GRANDES GRANULARES T (LLGG) LEUCEMIA AGRESIVA CÉLULAS NK . LEUCEMIA/LINFOMA T DEL ADULTO (HTLV-I+)
 LEUCEMIA A LINFOCITOS GRANDES GRANULARES T (LLGG) LEUCEMIA AGRESIVA CÉLULAS NK . LEUCEMIA/LINFOMA T DEL ADULTO (HTLV-I+)")
6
LINFOMAS LINFOMA DE HODGKIN LINFOMA NO HODGKIN

7
NEOPLASIAS LINFOIDES DE CELULAS MADURAS
Leucemias de origen B: 95% de los casos LLC constituye el 80% LPL-B el 10% Tricoleucemia el 10%. Leucemias de origen T: 5% de los casos

8
LEUCEMIA LINFOCITICA CRONICA
DEFINICIÓN. Enfermedad caracterizada por la proliferación y acumulación de Linfocitos B monoclonales de larga vida CD5+/CD19+ en sangre periférica, M.O, ganglios linfáticos y otros órganos linfoides secundarios, que han tenido contacto con el antígeno, morfológicamente tienen apariencia madura pero que son biológicamente inmaduros.

9
LEUCEMIA LINFOCITICA CRONICA
INCIDENCIA: 1-3 / habitantes/año. 20 a 30% de todas las leucemias. Especialmente en Occidente. SEXO: Relación hombre / mujer 2 a 1. EDAD DE PRESENTACIÓN: años, (media 55 años). ETIOLOGÍA: desconocida PATOLOGÍA MOLECULAR. Existen algunas asociaciones, pero no se ha encontrado oncogen ni alteraciones citogenéticas específicas.
. ETIOLOGÍA: desconocida. PATOLOGÍA MOLECULAR. Existen algunas asociaciones, pero no se ha encontrado oncogen ni alteraciones citogenéticas específicas.")
10
CLÍNICA Linfadenopatía, hepatoesplenomegalia.
A medida que la enfermedad evoluciona, pueden existir síntomas relacionados a plaquetopenia autoinmune, anemia hemolítica autoinmne o crisis de aplasia. El estadio clínico se establece en base a dos sistemas Rai o Binet. Los estadios 0 sólo tiene linfocitosis y va aumentando el score a medida que aparece organomegalia, anemia y/o trombocitopenia.

11
Diagnóstico Hemograma
Leucocitosis a veces muy ligeras (12000/mm3), rara vez llegan a 50000/mm3, con linfocitosis relativas de alrededor de 80%. No anemia , no plaquetopenia.
, rara vez llegan a 50000/mm3, con linfocitosis relativas de alrededor de 80%. No anemia , no plaquetopenia.")
12
DIAGNÓSTICO: Número de linfocitos mayor a 5000/l. (4 semanas)
Morfología: linfocitos pequeños, raramente pueden aparecer algunos linfocitos clivados. En el 15% de las LLC la morfología es atípica: ya sea debido a la presencia de más del 15% de L clivados o a la presencia de más del 10% de prolinfocitos típicos LLC/LPL Más de un 30% de linfocitos en la MO

13
MORFOLOGÍA

14
MORFOLOGÍA

15
MORFOLOGÍA

16
INMUNOFENOTIPO IgS de baja intensidad. (IgM – IgD) Expresión de CD 19, CD20 y CD23 Coexpresión del CD 5 (panT) FMC7 negativo. ZAP 70 CD 38 CD 49b

17
CITOGENÉTICA 25% deleción 13q14. Cercano al locus del gen supresor de retinoblastoma (Rb). Se deleciona un gen que codifica 2 micro RNAs Esos micro RNAs disminuyen la expresión de la proteína antiapoptótica bcl-2. Evolución favorable. 10-20% trisomía del 12. Se asocia generalmente a morfología atípica, actividad proliferativa alta, presencia de prolinfocitos, marcada leucocitosis y mal pronóstico. A veces esta anomalía aparece cuando progresa la enfermedad. 10-20% 11q-, deleción 11q Se observa en pacientes menores de 55 años. Faltaría un gen supresor de tumor, gen que tiene importancia en la activación del producto del gen de la proteína 53. Estos pacientes presentan grandes adenomegalias, y corta sobrevida. Estas células expresan niveles disminuidos de moléculas de adhesión.
. Se deleciona un gen que codifica 2 micro RNAs Esos micro RNAs disminuyen la expresión de la proteína antiapoptótica bcl-2. Evolución favorable % trisomía del 12. Se asocia generalmente a morfología atípica, actividad proliferativa alta, presencia de prolinfocitos, marcada leucocitosis y mal pronóstico. A veces esta anomalía aparece cuando progresa la enfermedad % 11q-, deleción 11q Se observa en pacientes menores de 55 años. Faltaría un gen supresor de tumor, gen que tiene importancia en la activación del producto del gen de la proteína 53. Estos pacientes presentan grandes adenomegalias, y corta sobrevida. Estas células expresan niveles disminuidos de moléculas de adhesión.")
18
mRNAs y Ciclo Celular

19
mRNAs y control de la Apoptosis

20
LEUCEMIA LINFOCITICA CRONICA
CLASIFICACION En base a la presencia de mutaciones somáticas de IgV(H), la LLC ha sido recientemente subdividida en 2 subgrupos: LLC-B no mutada o UCLL derivada de los LB ubicados en los centros pre germinales, de mal pronóstico. LLC-B de memoria o MCLL, de buen pronóstico
, la LLC ha sido recientemente subdividida en 2 subgrupos: LLC-B no mutada o UCLL derivada de los LB ubicados en los centros pre germinales, de mal pronóstico. LLC-B de memoria o MCLL, de buen pronóstico.")
21
LEUCEMIA LINFOCITICA CRONICA
Para reconocer al clon de linfocitos con mutación del gen IgVH se usan los marcadores: CD 38, ZAP-70, CD49d y el AICD. ZAP-70 es una proteína tipo tirosinquinasa que se puede medir por citometría de flujo En las ULLC hay aumento de ZAP-70 y CD 38. Las MCLL tienen disminución del ZAP-70 y CD 38

24
LEUCEMIA LINFOCITICA CRONICA
Los linfocitos de la LLC derivan de los LB activados y han sufrido una exposición al antígeno. Esto se evidencia por la presencia de marcadores de activación tales como el CD23, CD25, CD69 y CD71, tanto en variante la mutada como en la no mutada.

25
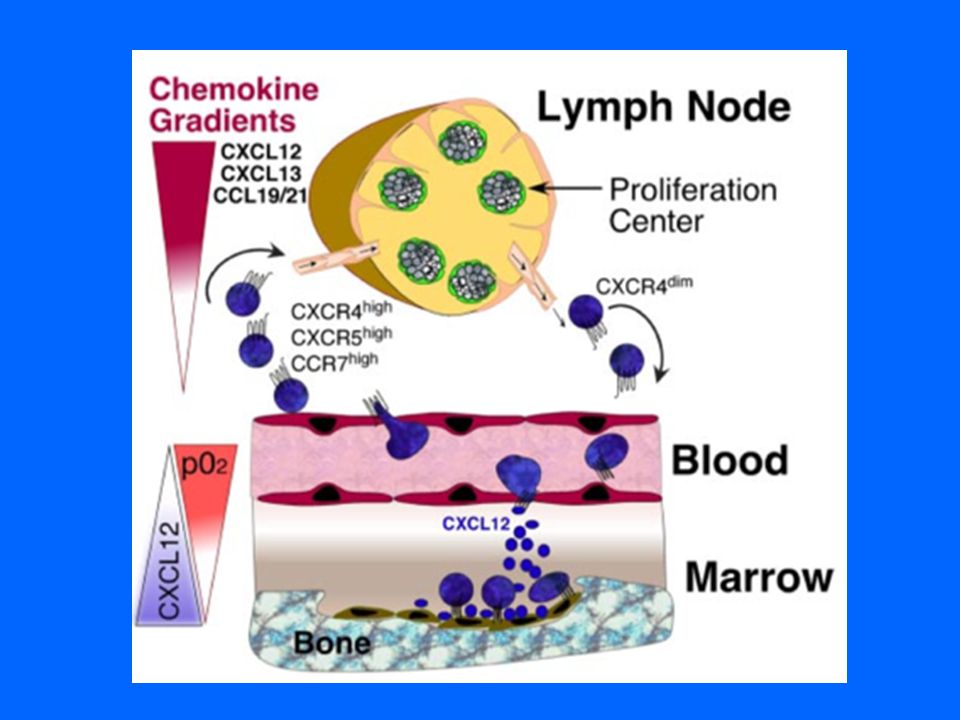
INTERACCIONES ENTRE EL MICROAMBIENTE Y APOPTOSIS.
Las células de la LLC tienen larga sobrevida in vivo lo que indica la importancia de la influencia del microambiente de los tejidos linfoides. El contacto con las NLC, BMSC, dendríticas y LT, genera un aumento de proteínas antiapoptóticas en el citoplasma de los linfocitos de la LLC y mejora la migración hacia los órganos linfoides y MO

27
LEUCEMIA LINFOCITICA CRONICA
A pesar de la existencia de los dos tipos de LLC todas derivan de los LB de memoria. Cuando se une un Ag al BCR del LB, si se trata de una LLC no mutada predomina una respuesta de activación y proliferación que se produce como consecuencia del aumento de la ZAP70 que interviene en la señalización. El aumento de CD 38 está directamente relacionado a esta respuesta.

28
LEUCEMIA LINFOCITICA CRONICA
En cambio, en las LLC mutadas predomina la respuesta anérgica y la antiapoptosis. Esto explica que en las mutadas prima la acumulación. Son CD5 dism y CXCR4 alto. En cambio las no mutadas, en las que prima la proliferación tienen una velocidad de duplicación linfocitaria alta y peor pronóstico.

29
CD 38 + CD 38 -

31
INTERACCIONES ENTRE EL MICROAMBIENTE Y APOPTOSIS.
Los LT CD4+ ayudan a atraer más células de LLC La interacción con el CD40L del LT, aumenta la síntesis de proteínas antiapoptóticas. Lo mismo ocurre cuando el CD 38 se une a su ligando sobre las NLC y aumenta la expresión de ZAP-70

32
EL ROL DE LOS LT EN LA LLC LT están incrementados y tanto los LT CD4+ como los CD8+ de la sangre periférica muestran oligoclonalidad. Existe aumento de los NK y LT supresores, A pesar del aumento, los NK tienen disminuida la producción y/o liberación de mediadores citolíticos solubles.

33
ALTERACIÓN DE LA INMUNIDAD CELULAR
Deficiencia inmune adquirida y susceptibilidad a infecciones por Herpes Zoster y asociación con Carcinoma cutáneo basocelular.

34
ALTERACIÓN DE LA INMUNIDAD HUMORAL
La Ig de superficie tiene capacidad de unión polirreactiva, reacciona contra sus propias Igs del plasma: IgM, IgG e IgA, produciendo hipogammaglobulinemia y por lo tanto inmunosupresión. Tiene alta prevalencia de fenómenos autoinmunes. El clon leucémico de la LLC comúnmente expresa IgM que muestra amplia reactividad contra Ags. propios.

35
FACTORES PRONÓSTICOS Buen pronóstico: Sexo: F
Estadío clínico: Binet A o Rai 0, 1. Infiltración de MO: no difusa Morfología : Típica Tiempo de duplicación de los L: > de 12m. CD38 >20-30% Anomalías citogenéticas: Ninguna ó 13q- Estado gen IgVH: Mutado ZAP 70: Bajo 2 microglobulina: baja CD23 soluble: Bajo

36
TRATAMIENTO Pacientes mayores de 65 años con enfermedad indolente no son tratados. Sólo cuando se tiene una idea clara de progresión se comienza con el mismo por vía oral. El transplante de M.O puede proponerse para aquellos que han llegado a remisión completa y muestran signos tempranos de recaída, preferentemente autotransplante.

38
LEUCEMIA PROLINFOCITICA B (LPL-B)
DEFINICIÓN: Es una neoplasia de células B caracterizada por la presencia de prolinfocitos en SP, M.O y bazo. Provienen de LB maduros. ETIOLOGÍA: desconocida. Puede provenir de una LLC.

39
LEUCEMIA PROLINFOCITICA B (LPL-B)
INCIDENCIA: 10 % de las Neoplasias L crónicas. RELACIÓN HOMBRE-MUJER: 4:1 EDAD DE PRESENTACIÓN: El 50 % de los pacientes tienen más de 70 años.

40
LEUCEMIA PROLINFOCITICA B (LPL-B)
MANIFESTACIONES CLÍNICAS: Esplenomegalia masiva, no adenopatías. Trastornos secundarios a la gran esplenomegalia. CITOGENETICA: 14q+, 6q-, trisomía del 12, (génicas): mutaciones en la p53.
: mutaciones en la p53.")
41
LEUCEMIA PROLINFOCITICA B (LPL-B)
HEMOGRAMA: Leucocitosis > de /l, con linfocitosis y prolinfocitos > del 55%. Generalmente hay anemia y ligera plaquetopenia. MÉDULA ÓSEA: Infiltrado del mismo tipo de células.

42
LEUCEMIA PROLINFOCITICA B

43
LEUCEMIA PROLINFOCITICA B

44
LEUCEMIA PROLINFOCITICA B (LPL-B)
INMUNOFENOTIPO. Ig de superficie fuerte, CD22, FMC-7 y CD79a positivos, CD5 débil, CD23 negativo. TERAPEÚTICA. Similar a la de la LLC. Mal pronóstico.

46
TRICOLEUCEMIA Hairy Cell Leukemia (HCL)
Definición: La leucemia de células vellosas es una neoplasia de linfocitos B que afecta fundamentalmente a la MO y al bazo. Se caracteriza por presentar células con prolongaciones citoplasmáticas prominentes

47
HCL Relación hombre : mujer: 4:1
Edad de presentación: mediana de 52 años Clínica: esplenomegalia masiva en el 90% de los pacientes. Adenopatías poco frecuentes.

48
HCL Hemograma: presenta pancitopenia periférica.
Linfocitosis relativa y neutropenia absoluta. Los linfocitos son pequeños con proyecciones citoplasmáticas delgadas, núcleo generalmente redondeado y, a veces, se puede apreciar un nucleolo. El citoplasma no posee granulaciones. Estas células pueden caracterizarse con microscopía electrónica o bien por pruebas citoquímicas.

49
HCL

50
HCL

51
HCL

52
HCL Médula ósea: se observa fibrosis asociada, pero se identifican fácilmente los tricoleucocitos mezclados con tejido hematopoyético residual. Reticulina + Citoquímica: el citoplasma de las células vellosas se tiñe habitualmente con la TRAP (fosfatasa ácida tartrato resistente). Si bien la fosfatasa ácida es característica de los LT, en ese caso se hace positiva a la isoenzima 5 que es resistente al tartrato.
. Si bien la fosfatasa ácida es característica de los LT, en ese caso se hace positiva a la isoenzima 5 que es resistente al tartrato.")
53
HCL- TRAP

54
HCL Inmunofenotipo: IgS fuerte, CD19, CD20, CD22 y CD25 positivos. El CD103 tiene la mayor sensibilidad y especificidad para estos procesos Evolución y pronóstico: con la terapia actual la tasa de sobrevida es del 95% a los 4 años.

56
LEUCEMIA PROLINFOCÍTICA T (LPLT)
DEFINICIÓN: enfermedad caracterizada por la proliferación de prolinfocitos de origen T. INCIDENCIA: menos del 5% de todas las NLC, pero el 30 % de todas las neoplasias T. RELACIÓN HOMBRE-MUJER: 3:2

57
LEUCEMIA PROLINFOCÍTICA T (LPLT)
MANIFESTACIONES CLÍNICAS: -Esplenomegalia. -Manifestaciones cutáneas en torso, brazos y cara: eritema difuso infiltrado, erupción papular no descamativa y no pruriginosa. -Derrames pleurales, ascitis y compromiso del SNC.

58
LEUCEMIA PROLINFOCÍTICA T (LPLT)
HEMOGRAMA: Linfocitosis variable de a /l. Se observan abundantes prolinfocitos, de núcleo cerebriforme, citoplasma celeste sin granulaciones. Generalmente tiene un nucleolo a veces menos visible que en la LPLB, el tamaño es algo menor y el citoplasma más basófilo. MÉDULA ÓSEA Y PIEL: se encuentran infiltradas por el mismo tipo de células.

59
LEUCEMIA PROLINFOCÍTICA T (LPLT)
")
60
LEUCEMIA PROLINFOCÍTICA T (LPLT)
")
61
LEUCEMIA PROLINFOCÍTICA T (LPLT)
INMUNOFENOTIPO: Expresan un fenotipo postímico con expresión de marcadores pan T: CD2, CD4, CD5, CD7 y CD 26 y CD 52 positivos. Algunas expresan CD4 pero no CD8, a veces expresan tanto CD4 como CD 8 (células más inmaduras). Algunos expresan el TCR-/ y TCL-1 .
. Algunos expresan el TCR-/ y TCL-1 .")
62
LEUCEMIA PROLINFOCÍTICA T (LPLT)
CITOQUÍMICA: fosfatasa ácida y beta glucuronidasa positiva CURSO CLÍNICO AGRESIVO Y MAL PRONÓSTICO

63
LLC LPL-B HCL LPL-T Leucocitos ˃ Pancitopenia FLR ↑ Linfocitos ˃ 55% Prol ↓N Abs ↑L Rel ↑ Prol Clínica Adenomegalia Hepatomegalia Esplenomegalia No Adenomeg. Esplenomeg. Infiltrado de Piel Edad 50 – 75 ˃ 70 ≈ 52 Incidencia 80% NLPc 10% NLPc ˂ 5% NLPc Fenotipo IgS débil CD19 CD20 CD27 CD23 CD5 débil FMC7 (-) IgS Fuerte CD22 CD23 (-) FMC7 (+) CD25 CD103 CD2 CD4 CD5 CD7 CD26 TCR – TCL1 Pronóstico Moderado Malo
 IgS Fuerte. CD22. CD23 (-) FMC7 (+) CD25. CD103. CD2. CD4. CD5. CD7. CD26. TCR – TCL1. Pronóstico. Moderado. Malo.")
64
GRACIAS

Presentaciones similares














, bcl2 s Ig, CD19,CD20,CD21 SOBREVIDA 7-9 AÑOS TRASFORMACION.>")










