Descargar la presentación
La descarga está en progreso. Por favor, espere

1
Daniela González Largo

2
Enfrentamiento Paciente Parkinsoniano
Enfermedad de Parkinson (PD) Parkinsonismos Atípicos Parálisis supranuclear progresiva (PSP) Degeneración corticobasal (DCB) Atrofia multisistémica (AMS) “Atípicos” Parkinsonismos Atípicos
 Parkinsonismos Atípicos. Parálisis supranuclear progresiva (PSP) Degeneración corticobasal (DCB) Atrofia multisistémica (AMS) Atípicos Parkinsonismos Atípicos.")
3
Parkinsonismos Genéticos
Nuevos parkinsonismos han emergido debido a avances en la genética Pueden compartir algunas características clínicas con los fenotipos clásicos de PSP, DCB y AMS (imitadores) En Parkinsonismos atípicos “clásicos” Diagnóstico definitivo requiere confirmación patológica Mal pronóstico No hay tratamientos disponibles Algunas condiciones genéticas pueden diagnosticarse in vivo mediante pruebas genéticas y posibilidades terapeuticas
 En Parkinsonismos atípicos clásicos Diagnóstico definitivo requiere confirmación patológica. Mal pronóstico. No hay tratamientos disponibles. Algunas condiciones genéticas pueden diagnosticarse in vivo mediante pruebas genéticas y posibilidades terapeuticas.")
4
Sospecha Parkinsonismos “Like”
Ciertas características clínicas Antecedentes familiares Edad más temprana de inicio

5
Dificultades Diagnóstico Diferencial
Pacientes con diagnóstico histopatológico se presentan fenotípicamente no “puros” Errores diagnósticos frecuentes con Demencia por cuerpos de Lewy, Demencia Frontotemporal y Enfermedad de Alzheimer Parkinsonismos secundarios a enfermedades vasculares, infecciosas, inducida por fármacos, enfermedades autoinmunes y trastornos paraneoplásicos deben descartarse previamente inicio agudo o subagudo, antecedente de abuso de drogas o factores de riesgo vascular, signos sugestivos de proceso infeccioso, y examen de imágenes por resonancia magnética [RM] y el líquido cefalorraquídeo [LCR]

7
Características Subgrupos
PSP-símil DCB-símil AMS-símil Parkinsonismo Alteraciones oculomotoras tempranas Inestabilidad postural precoz con caídas Deterioro frontal precoz Parkinsonismo asimétrico Signos corticales: Apraxia, Mano alienada Mioclonías corticales Pérdida sensorial cortical Parkinsonismo y disfunción autonómica precoz o Parkinsonismo y síndrome cerebeloso PSP: lentitud de movimientos sacádicos verticales

8
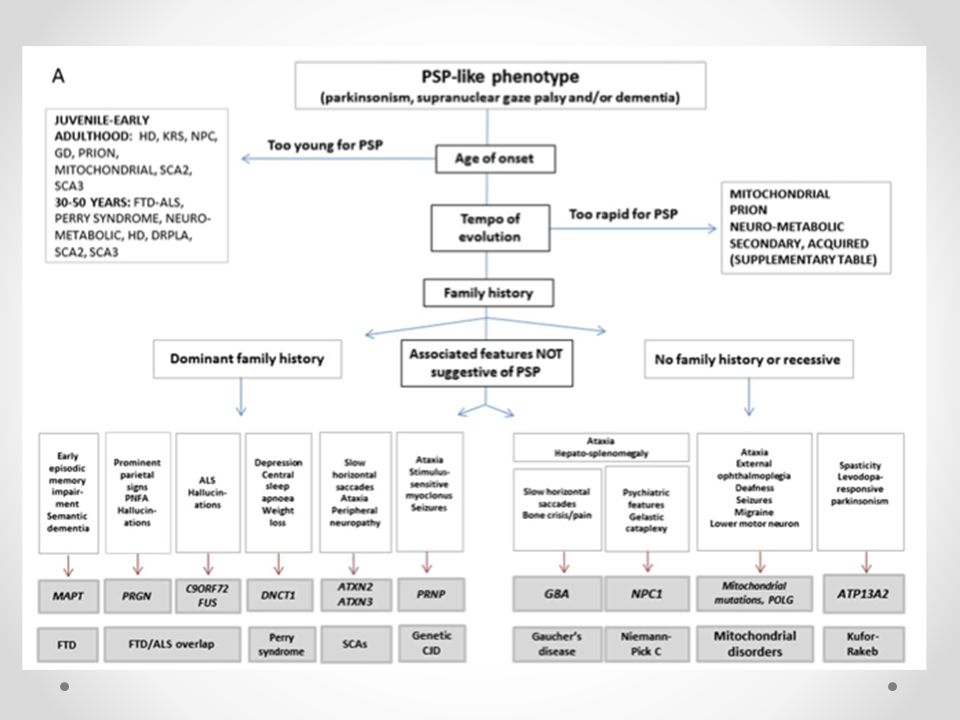
PSP-símil Genéticas Neurodegenerativas Neurometabólicas Priónicas
Degeneración Frontotemporal Síndrome de Perry Síndrome Kufor-Rakeb Neurometabólicas Niemann-Pick C Enfermedad de Gaucher Trastornos Mitocondriales Priónicas Steele, Richardson y Olszewski describieron esta enfermedad en 1964, también conocida como Parálisis Supranuclear Progresiva (PSP). Se caracteriza por trastornos de la mirada conjugada vertical, especialmente hacia abajo, parálisis pseudobulbar, disartria, distonia axial y demencia, Tb blefarospasm y retrocolis. Después de los cuarenta años y, en la mayoría de los casos, alrededor de los sesenta años. Trastornos motores que se presentan como un cuadro de parkinsonismo acinético rígido asociado a una pérdida de los reflejos posturales con caídas frecuentes que aparecen dentro del primer año de evolución, lo cual lleva a la imposibilidad de mantenerse en pie. Los pacientes presentan un aumento del tono muscular axial respecto al distal con extensión del cuello en retrocollis, adoptando una postura erecta en hiperextensión, que le da una apariencia de majestuosidad (distonia axial). El trastorno de la mirada se caracteriza por un enlentecimiento y, finalmente, la imposibilidad de ejecutar los movimientos oculares en especial en la mirada vertical conjugada hacia abajo. En el adulto mayor con frecuencia se pueden encontrar trastornos de la mirada conjugada hacia arriba y de la convergencia que no tienen significado patológico. El trastorno de la mirada conjugada observado en la PSP está presente en los movimientos voluntarios y de seguimiento, sin embargo, esta limitación es menor en los movimientos oculares reflejos, como al obtener el reflejo oculocefálico, en que se le gira activamente la cabeza al paciente y se obtiene el fenómeno conocido como “ojos de muñecas”. Con frecuencia este reflejo no es explorado en el plano vertical. Estos trastornos son de presentación gradual pudiendo llegar con la evolución de la enfermedad a un oftalmoplejía, en que se compromete la mirada conjugada en todos los sentidos e incluso en un pequeño número de pacientes se puede perder la respuesta refleja. En la primera etapa de la enfermedad se compromete la velocidad de los movimientos sacádicos de los ojos. En muchos casos se presenta una hipometría y descomposición de los movimientos sacádicos, similar al fenómeno conocido en la literatura anglosajona como “square wave jerks”. Se puede asociar a blefaroespasmo que, en algunos casos, se comporta como una apraxia parpebral, es decir, una dificultad para abrir voluntariamente los ojos por la inhibición supranuclear del músculo elevador de los párpados. Varias enfermedades neurológicas se presentan con trastornos de la mirada (adaptado de Lees A,1995): Atrofia Multisistémica tipo olivopontocerebelosa. Degeneración Corticobasal. Atrofia Dento-palido-nigro-lusial. Niemann-Pick de inicio tardío. Enfermedad de Joseph. Los trastornos conductuales y neuropsiquiátricos, más frecuentes son el síndrome frontal, se presentan como una falla de la función ejecutiva, apatía, cambio de personalidad, prensión forzada, perseveración de las conductas, enlentecimiento de pensamiento, disminución de la atención, disminución de la fluencia verbal. En muchos casos, se constituye un cuadro de demencia de tipo fronto-límbico. El síndrome pseudobulbar está presente, en todos los pacientes, en diferentes grados y puede ser uno de los síntomas de inicio de la enfermedad. Se presenta disartria que evoluciona a anartria. Trastornos de la deglución, que llegan a requerir uso de gastrostomía para la nutrición, siendo un factor de mal pronóstico su aparición precoz. También se presentan otros síntomas y signos de liberación piramidal como la risa y llanto espasmódico, reflejo de Babinski, exaltación de reflejos osteotendíneos. La PSP se presenta habitualmente en la séptima década de la vida. Es un cuadro que progresa rápidamente con importante trastornos del equilibrio, inestabilidad de la marcha y caídas frecuentes, lo cual suele ser el primer síntoma. Durante el primer año de evolución, se asocian disartria y un síndrome acineto-rígido que no responde al tratamiento con levodopa. En la mitad de los casos durante el primer año de evolución aparecen los trastornos conductuales, sin embargo, es poco frecuente que éstos sean los primeros síntomas. Los trastornos de la mirada conjugada vertical, que son característicos de la enfermedad, están presentes durante el primer año en más de la mitad de los casos, pero en muchos de ellos pueden aparecer en estados más tardíos de la enfermedad. La sobrevida media desde el inicio de la enfermedad es de aproximadamente 5 a 7 años. Aquellos pacientes que presentan caídas frecuentes durante el primer año de evolución tienen una sobrevida más corta. La causa de muerte más frecuente son los cuadro broncopulmonares secundarios a aspiración. El diagnóstico diferencial con los otros síndromes acineto-rígidos, como la enfermedad de Parkinson, atrofia multisistémica, degeneración córtico basal, demencia de cuerpos de Lewy, enfermedad de Alzheimer, enfermedad cerebrovascular multinfarto, habitualmente es muy difícil, especialmente, en las etapas iniciales de estas enfermedades. Los criterios clínicos de diagnóstico no permiten certeza. Criterios clínicos para el diagnóstico de la PSP: Criterios mayores de inclusión Enfermedad gradualmente progresiva. Inicio después de los 40 años Trastorno de la mirada vertical de tipo supranuclear Inestabilidad postural, con caídas frecuentes de aparición precoz Criterios de apoyo • Síndrome acineto-rígido simétrico con rigidez de predominio proximal. • Postura de cuello anormal (retrocollis). • Pobre o ausente respuesta del parkinsonismo al uso de levodopa o agonistas dopaminérgicos. • Disfagia y disartria de inicio precoz. • Trastorno cognitivo de tipo frontal con inicio precoz.
. Se caracteriza por trastornos de la mirada conjugada vertical, especialmente hacia abajo, parálisis pseudobulbar, disartria, distonia axial y demencia, Tb blefarospasm y retrocolis. Después de los cuarenta años y, en la mayoría de los casos, alrededor de los sesenta años. Trastornos motores que se presentan como un cuadro de parkinsonismo acinético rígido asociado a una pérdida de los reflejos posturales con caídas frecuentes que aparecen dentro del primer año de evolución, lo cual lleva a la imposibilidad de mantenerse en pie. Los pacientes presentan un aumento del tono muscular axial respecto al distal con extensión del cuello en retrocollis, adoptando una postura erecta en hiperextensión, que le da una apariencia de majestuosidad (distonia axial). El trastorno de la mirada se caracteriza por un enlentecimiento y, finalmente, la imposibilidad de ejecutar los movimientos oculares en especial en la mirada vertical conjugada hacia abajo. En el adulto mayor con frecuencia se pueden encontrar trastornos de la mirada conjugada hacia arriba y de la convergencia que no tienen significado patológico. El trastorno de la mirada conjugada observado en la PSP está presente en los movimientos voluntarios y de seguimiento, sin embargo, esta limitación es menor en los movimientos oculares reflejos, como al obtener el reflejo oculocefálico, en que se le gira activamente la cabeza al paciente y se obtiene el fenómeno conocido como ojos de muñecas . Con frecuencia este reflejo no es explorado en el plano vertical. Estos trastornos son de presentación gradual pudiendo llegar con la evolución de la enfermedad a un oftalmoplejía, en que se compromete la mirada conjugada en todos los sentidos e incluso en un pequeño número de pacientes se puede perder la respuesta refleja. En la primera etapa de la enfermedad se compromete la velocidad de los movimientos sacádicos de los ojos. En muchos casos se presenta una hipometría y descomposición de los movimientos sacádicos, similar al fenómeno conocido en la literatura anglosajona como square wave jerks . Se puede asociar a blefaroespasmo que, en algunos casos, se comporta como una apraxia parpebral, es decir, una dificultad para abrir voluntariamente los ojos por la inhibición supranuclear del músculo elevador de los párpados. Varias enfermedades neurológicas se presentan con trastornos de la mirada (adaptado de Lees A,1995): Atrofia Multisistémica tipo olivopontocerebelosa. Degeneración Corticobasal. Atrofia Dento-palido-nigro-lusial. Niemann-Pick de inicio tardío. Enfermedad de Joseph. Los trastornos conductuales y neuropsiquiátricos, más frecuentes son el síndrome frontal, se presentan como una falla de la función ejecutiva, apatía, cambio de personalidad, prensión forzada, perseveración de las conductas, enlentecimiento de pensamiento, disminución de la atención, disminución de la fluencia verbal. En muchos casos, se constituye un cuadro de demencia de tipo fronto-límbico. El síndrome pseudobulbar está presente, en todos los pacientes, en diferentes grados y puede ser uno de los síntomas de inicio de la enfermedad. Se presenta disartria que evoluciona a anartria. Trastornos de la deglución, que llegan a requerir uso de gastrostomía para la nutrición, siendo un factor de mal pronóstico su aparición precoz. También se presentan otros síntomas y signos de liberación piramidal como la risa y llanto espasmódico, reflejo de Babinski, exaltación de reflejos osteotendíneos. La PSP se presenta habitualmente en la séptima década de la vida. Es un cuadro que progresa rápidamente con importante trastornos del equilibrio, inestabilidad de la marcha y caídas frecuentes, lo cual suele ser el primer síntoma. Durante el primer año de evolución, se asocian disartria y un síndrome acineto-rígido que no responde al tratamiento con levodopa. En la mitad de los casos durante el primer año de evolución aparecen los trastornos conductuales, sin embargo, es poco frecuente que éstos sean los primeros síntomas. Los trastornos de la mirada conjugada vertical, que son característicos de la enfermedad, están presentes durante el primer año en más de la mitad de los casos, pero en muchos de ellos pueden aparecer en estados más tardíos de la enfermedad. La sobrevida media desde el inicio de la enfermedad es de aproximadamente 5 a 7 años. Aquellos pacientes que presentan caídas frecuentes durante el primer año de evolución tienen una sobrevida más corta. La causa de muerte más frecuente son los cuadro broncopulmonares secundarios a aspiración. El diagnóstico diferencial con los otros síndromes acineto-rígidos, como la enfermedad de Parkinson, atrofia multisistémica, degeneración córtico basal, demencia de cuerpos de Lewy, enfermedad de Alzheimer, enfermedad cerebrovascular multinfarto, habitualmente es muy difícil, especialmente, en las etapas iniciales de estas enfermedades. Los criterios clínicos de diagnóstico no permiten certeza. Criterios clínicos para el diagnóstico de la PSP: Criterios mayores de inclusión. Enfermedad gradualmente progresiva. Inicio después de los 40 años. Trastorno de la mirada vertical de tipo supranuclear. Inestabilidad postural, con caídas frecuentes de aparición precoz. Criterios de apoyo. • Síndrome acineto-rígido simétrico con rigidez de predominio proximal. • Postura de cuello anormal (retrocollis). • Pobre o ausente respuesta del parkinsonismo al uso de levodopa o agonistas dopaminérgicos. • Disfagia y disartria de inicio precoz. • Trastorno cognitivo de tipo frontal con inicio precoz.")
9
PSP-símil Neurodegenerativas
Degeneración Frontotempral Variante de comportamiento ( bvFTD ) Heredadas en un patrón dominante en gen de proteina tau y de progranulina (TDP-43) Estudios para secuenciación para tau asociada a microtúbulos ( MAPT ) y progranulina ( PGRN ) o niveles plasmáticos progranulina Mutaciones MAPT: 3ra al 5ta década, media 40 años, (25-65 años) Historia familiar positiva de parkinsonismo o demencia (penetración casi del 100%) Deterioro de la memoria episódica y demencia semántica Problemas conductuales suelen preceder a los signos clásicos de PSP (20%) RM: PSP muestra atrofia mesencéfalo y diencéfalo y MAPT pueden mostrar una atrofia frontotemporal simétrica DaTSCAN es anormal en ambos y escasa respuesta a levodopa ioflupane iodine-123 injection or DaTscan, a contrast agent to be used with single-photon emission computed tomography (SPECT) for detecting dopamine transporters (DaT) in suspected parkinsonian syndromes. Mutaciones asociada a la proteína tau y progranulina [TDP- 43] , mientras que las mutaciones ocurren raramente MAPT en casos esporádicos. Con respecto a las características asociadas
 Heredadas en un patrón dominante en gen de proteina tau y de progranulina (TDP-43) Estudios para secuenciación para tau asociada a microtúbulos ( MAPT ) y progranulina ( PGRN ) o niveles plasmáticos progranulina. Mutaciones MAPT: 3ra al 5ta década, media 40 años, (25-65 años) Historia familiar positiva de parkinsonismo o demencia (penetración casi del 100%) Deterioro de la memoria episódica y demencia semántica. Problemas conductuales suelen preceder a los signos clásicos de PSP (20%) RM: PSP muestra atrofia mesencéfalo y diencéfalo y MAPT pueden mostrar una atrofia frontotemporal simétrica. DaTSCAN es anormal en ambos y escasa respuesta a levodopa. ioflupane iodine-123 injection or DaTscan, a contrast agent to be used with single-photon emission computed tomography (SPECT) for detecting dopamine transporters (DaT) in suspected parkinsonian syndromes. Mutaciones asociada a la proteína tau y progranulina [TDP- 43] , mientras que las mutaciones ocurren raramente MAPT en casos esporádicos. Con respecto a las características asociadas.")
10
PSP-símil Neurodegenerativas
Degeneración Frontotempral Mutaciones PGRN La edad media de inicio 60 años (35-83 años ) Penetrancia del 90 % a la edad de 70 años (antecedente no siempre presente) y esporádico en 3% de DFT Disfunción frontal mucho antes del cuadro PSP-símil Parálisis supranuclear de la mirada Afectación del lóbulo parietal (discalculia , apraxia Alucinaciones (25 %) RM atrofia frontotemporoparietal asimétrica Mutación del gen C9ORF72 (TDP-43) síndrome de superposición esclerosis lateral amiotrófica-DFT Se inician como PSP en el 35% signos compromiso neurona motora superior o inferior Presencia de alucinaciones ioflupane iodine-123 injection or DaTscan, a contrast agent to be used with single-photon emission computed tomography (SPECT) for detecting dopamine transporters (DaT) in suspected parkinsonian syndromes.
 Penetrancia del 90 % a la edad de 70 años (antecedente no siempre presente) y esporádico en 3% de DFT. Disfunción frontal mucho antes del cuadro PSP-símil. Parálisis supranuclear de la mirada. Afectación del lóbulo parietal (discalculia , apraxia. Alucinaciones (25 %) RM atrofia frontotemporoparietal asimétrica. Mutación del gen C9ORF72 (TDP-43) síndrome de superposición esclerosis lateral amiotrófica-DFT. Se inician como PSP en el 35% signos compromiso neurona motora superior o inferior. Presencia de alucinaciones. ioflupane iodine-123 injection or DaTscan, a contrast agent to be used with single-photon emission computed tomography (SPECT) for detecting dopamine transporters (DaT) in suspected parkinsonian syndromes.")
11
PSP-símil Neurodegenerativas
Sindrome de Perry Autosómico dominante infrecuente Mutaciones en el gen dynactin ( DCTN1 ) con inclusionesTDP- 43 Edad de inicio 30 a 61 años (media 45 años) Penetrancia es cercana al 50 % Fenotipo incluye parkinsonismo con diferentes combinaciones de hipoventilación central , pérdida de peso extrema y síntomas psiquiátricos como atimormia (aquinesia síquica), apatía y alucinaciones Respuesta a la L -dopa varía desde ninguna respuesta a la mejora y el desarrollo significativo de las fluctuaciones motoras y discinesias Historia familiar puede ser positivo para los signos de hipoventilación central, muerte súbita durante el sueño en " no afectados ” RM y DaTSCAN es anormal tanto en el síndrome de Perry y PSP Atrofia mesencefalo
 con inclusionesTDP- 43. Edad de inicio 30 a 61 años (media 45 años) Penetrancia es cercana al 50 % Fenotipo incluye parkinsonismo con diferentes combinaciones de hipoventilación central , pérdida de peso extrema y síntomas psiquiátricos como atimormia (aquinesia síquica), apatía y alucinaciones. Respuesta a la L -dopa varía desde ninguna respuesta a la mejora y el desarrollo significativo de las fluctuaciones motoras y discinesias. Historia familiar puede ser positivo para los signos de hipoventilación central, muerte súbita durante el sueño en no afectados RM y DaTSCAN es anormal tanto en el síndrome de Perry y PSP. Atrofia mesencefalo.")
12
PSP-símil Neurodegenerativas
Síndrome Kufor-Rakeb Autosómico recesivo raro Mutaciones en el gen ATP13A2 , que codifica una ATPasa lisosomal 5 de tipo P (PARK9) El gen se identificó en una familia de Chile Inicio juvenil (12-29 años) Parkinsonismo, paralisis mirada vertical, disfunción cognitiva ( demencia y alucinaciones visuales) Además signos piramidales, espasmos distónicos oculógiras y mini- mioclono facial-fauces-dedos Parkinsonismo sensible a L- dopa (con fluctuaciones y discinesias ) RM: T2 ponderado: acumulación de hierro cerebral , y desde entonces nuevas mutaciones se han identificado en todo el mundo . No existe una patología de un paciente con síndrome de Kufor - Rakeb reportado , pero recientemente exoma secuenciación en una familia con patológicamente confirmado lipofuscinosis ceroide neuronal ha identificado mutaciones ATP13A2 que segregaban a los miembros de la familia afectados. en algunos pacientes , lo que puede ser una pista útil para sospechar este trastorno. La acumulación de hierro en T2 * pondera - RM es típico en la neurodegeneración con acumulación de hierro síndromes cerebro, pero también se ha reportado en adultos EP de aparición esporádica y síndromes parkinsonianos atípicos, incluyendo PSP y MSA. Sin embargo , la diferente distribución del hierro en estos trastornos y el cuadro clínico proporcionar claves importantes para el diagnóstico diferencial .
 El gen se identificó en una familia de Chile. Inicio juvenil (12-29 años) Parkinsonismo, paralisis mirada vertical, disfunción cognitiva ( demencia y alucinaciones visuales) Además signos piramidales, espasmos distónicos oculógiras y mini- mioclono facial-fauces-dedos. Parkinsonismo sensible a L- dopa (con fluctuaciones y discinesias ) RM: T2 ponderado: acumulación de hierro cerebral. , y desde entonces nuevas mutaciones se han identificado en todo el mundo . No existe una patología de un paciente con síndrome de Kufor - Rakeb reportado , pero recientemente exoma secuenciación en una familia con patológicamente confirmado lipofuscinosis ceroide neuronal ha identificado mutaciones ATP13A2 que segregaban a los miembros de la familia afectados. en algunos pacientes , lo que puede ser una pista útil para sospechar este trastorno. La acumulación de hierro en T2 * pondera - RM es típico en la neurodegeneración con acumulación de hierro síndromes cerebro, pero también se ha reportado en adultos EP de aparición esporádica y síndromes parkinsonianos atípicos, incluyendo PSP y MSA. Sin embargo , la diferente distribución del hierro en estos trastornos y el cuadro clínico proporcionar claves importantes para el diagnóstico diferencial .")
13
PSP-símil Neurometabólicas
Niemann-Pick C (mutaciones genes NPC ) Trastorno autosómica recesivo del almacenamiento de lípidos lisosomal, caracterizada por la acumulación de colesterol no esterificado y glicolípidos en el sistema endosomal/lisosomal. Es raro que se presente en adultos, segunda a tercera décadas en la mayoría de los pacientes ( hasta 54 años) Se pueden presentar de forma muy variada, paralisis mirada vertical (75%), ataxia cerebelosa, disartria, disfagia, disfunción cognitiva y síntomas psiquiátricos. Signos y síntomas viscerales incluyen esplenomegalia ( 54 %) con o sin hepatomegalia RM puede mostrar atrofia mesencéfalo. DaTSCAN no se han realizado Diagnóstico es hecho por tinción con filipina de fibroblastos de piel cultivados , con la posterior confirmación del diagnóstico realizado por análisis de mutaciones del NPC1 (la mayoría) y los genes NPC2 Miglustat es el único tratamiento aprobado, mejor resultado si es dado precoz Diagnóstico bioquímico de Niemann -Pick C es hecho por tinción con filipina de fibroblastos de piel cultivados , con la posterior confirmación del diagnóstico realizado por análisis de mutaciones del NPC1 (la mayoría) y los genes NPC2 . Además de la acumulación de colesterol , la enfermedad de NPC se neuropatológicamente caracteriza por la presencia de tau - positivo ovillos neurofibrilares , gliosis , desmielinización y pérdida de neuronas en regiones cerebrales seleccionadas .
 Trastorno autosómica recesivo del almacenamiento de lípidos lisosomal, caracterizada por la acumulación de colesterol no esterificado y glicolípidos en el sistema endosomal/lisosomal. Es raro que se presente en adultos, segunda a tercera décadas en la mayoría de los pacientes ( hasta 54 años) Se pueden presentar de forma muy variada, paralisis mirada vertical (75%), ataxia cerebelosa, disartria, disfagia, disfunción cognitiva y síntomas psiquiátricos. Signos y síntomas viscerales incluyen esplenomegalia ( 54 %) con o sin hepatomegalia. RM puede mostrar atrofia mesencéfalo. DaTSCAN no se han realizado. Diagnóstico es hecho por tinción con filipina de fibroblastos de piel cultivados , con la posterior confirmación del diagnóstico realizado por análisis de mutaciones del NPC1 (la mayoría) y los genes NPC2. Miglustat es el único tratamiento aprobado, mejor resultado si es dado precoz. Diagnóstico bioquímico de Niemann -Pick C es hecho por tinción con filipina de fibroblastos de piel cultivados , con la posterior confirmación del diagnóstico realizado por análisis de mutaciones del NPC1 (la mayoría) y los genes NPC2 . Además de la acumulación de colesterol , la enfermedad de NPC se neuropatológicamente caracteriza por la presencia de tau - positivo ovillos neurofibrilares , gliosis , desmielinización y pérdida de neuronas en regiones cerebrales seleccionadas .")
14
PSP-símil Neurometabólicas
Enfermedad de Gaucher (mutación del gen glucocerebrosidasa GBA ) Enfermedad de depósito lisosomal autosómica recesiva causada por mutaciones en el que conduce a la deficiencia de la enzima b - glucosidasa Se divide en 3 tipos: Gaucher 1 y 3 da parkinsonismo del adulto (3 y 5 década) Pacientes suelen tener movimientos sacádicos lentos y aumento de latencia de mirada horizontal (habitual DCB). Algunos con lentitud prominente de las sacadas verticales y disfunción cognitiva similar a PSP. Otras características neurológicas como caída de la cabeza ( 55 %) , ataxia ( 20 % ), convulsiones (16%) y la espasticidad ( 15 % ) Síntomas sistémicas como la esplenomegalia , hepatomegalia, dolor óseo, anemia y trombocitopenia Estado heterocigoto es el factor de riesgo genético más fuerte para la enfermedad de Parkinson esporádica ( 4 % a 5 %) Diagnóstico: medición de la actividad de GBA en los leucocitos ( baja) y citotriosidasa en plasma ( alto) y lpruebas posteriores del gen de GBA Patología muestra cuerpos de Lewy tronco cerebral y " células de Gaucher “ Terapia enzimática de sustitución ( alglucerasa , imiglucerasa ) y terapia de reducción de sustrato con miglustat . Es más frecuente en los Judios Ashkenazi. PSP en el que la latencia sacádicos es normal, y las sacadas verticales son más afectadas que los principios y horizontal , sobre todo a la baja
 Enfermedad de depósito lisosomal autosómica recesiva causada por mutaciones en el que conduce a la deficiencia de la enzima b - glucosidasa. Se divide en 3 tipos: Gaucher 1 y 3 da parkinsonismo del adulto (3 y 5 década) Pacientes suelen tener movimientos sacádicos lentos y aumento de latencia de mirada horizontal (habitual DCB). Algunos con lentitud prominente de las sacadas verticales y disfunción cognitiva similar a PSP. Otras características neurológicas como caída de la cabeza ( 55 %) , ataxia ( 20 % ), convulsiones (16%) y la espasticidad ( 15 % ) Síntomas sistémicas como la esplenomegalia , hepatomegalia, dolor óseo, anemia y trombocitopenia. Estado heterocigoto es el factor de riesgo genético más fuerte para la enfermedad de Parkinson esporádica ( 4 % a 5 %) Diagnóstico: medición de la actividad de GBA en los leucocitos ( baja) y citotriosidasa en plasma ( alto) y lpruebas posteriores del gen de GBA. Patología muestra cuerpos de Lewy tronco cerebral y células de Gaucher Terapia enzimática de sustitución ( alglucerasa , imiglucerasa ) y terapia de reducción de sustrato con miglustat. . Es más frecuente en los Judios Ashkenazi. PSP en el que la latencia sacádicos es normal, y las sacadas verticales son más afectadas que los principios y horizontal , sobre todo a la baja.")
15
PSP-símil Neurometabólicas
Trastornos Mitocondriales Mutaciones específicas (microdeleciones y deleciones) y también mutaciones polimerasa - gamma ( POLG ) heredadas en el modo dominante Pacientes PSP- símil (parkinsonismo, parálisis mirada vertical y disfunción cognitiva precoz) alrededor de los 60 años Sordera, ataxia y signos de neurona motora inferior Ptosis y oftalmoplejía infranuclear, migraña y epilepsia Excelente respuesta a la L- dopa Steele, Richardson y Olszewski describieron esta enfermedad en 1964, también conocida como Parálisis Supranuclear Progresiva (PSP). Se caracteriza por trastornos de la mirada conjugada vertical, especialmente hacia abajo, parálisis pseudobulbar, disartria, distonia axial y demencia, Tb blefarospasm y retrocolis. Después de los cuarenta años y, en la mayoría de los casos, alrededor de los sesenta años. Trastornos motores que se presentan como un cuadro de parkinsonismo acinético rígido asociado a una pérdida de los reflejos posturales con caídas frecuentes que aparecen dentro del primer año de evolución, lo cual lleva a la imposibilidad de mantenerse en pie. Los pacientes presentan un aumento del tono muscular axial respecto al distal con extensión del cuello en retrocollis, adoptando una postura erecta en hiperextensión, que le da una apariencia de majestuosidad (distonia axial). El trastorno de la mirada se caracteriza por un enlentecimiento y, finalmente, la imposibilidad de ejecutar los movimientos oculares en especial en la mirada vertical conjugada hacia abajo. En el adulto mayor con frecuencia se pueden encontrar trastornos de la mirada conjugada hacia arriba y de la convergencia que no tienen significado patológico. El trastorno de la mirada conjugada observado en la PSP está presente en los movimientos voluntarios y de seguimiento, sin embargo, esta limitación es menor en los movimientos oculares reflejos, como al obtener el reflejo oculocefálico, en que se le gira activamente la cabeza al paciente y se obtiene el fenómeno conocido como “ojos de muñecas”. Con frecuencia este reflejo no es explorado en el plano vertical. Estos trastornos son de presentación gradual pudiendo llegar con la evolución de la enfermedad a un oftalmoplejía, en que se compromete la mirada conjugada en todos los sentidos e incluso en un pequeño número de pacientes se puede perder la respuesta refleja. En la primera etapa de la enfermedad se compromete la velocidad de los movimientos sacádicos de los ojos. En muchos casos se presenta una hipometría y descomposición de los movimientos sacádicos, similar al fenómeno conocido en la literatura anglosajona como “square wave jerks”. Se puede asociar a blefaroespasmo que, en algunos casos, se comporta como una apraxia parpebral, es decir, una dificultad para abrir voluntariamente los ojos por la inhibición supranuclear del músculo elevador de los párpados. Varias enfermedades neurológicas se presentan con trastornos de la mirada (adaptado de Lees A,1995): Atrofia Multisistémica tipo olivopontocerebelosa. Degeneración Corticobasal. Atrofia Dento-palido-nigro-lusial. Niemann-Pick de inicio tardío. Enfermedad de Joseph. Los trastornos conductuales y neuropsiquiátricos, más frecuentes son el síndrome frontal, se presentan como una falla de la función ejecutiva, apatía, cambio de personalidad, prensión forzada, perseveración de las conductas, enlentecimiento de pensamiento, disminución de la atención, disminución de la fluencia verbal. En muchos casos, se constituye un cuadro de demencia de tipo fronto-límbico. El síndrome pseudobulbar está presente, en todos los pacientes, en diferentes grados y puede ser uno de los síntomas de inicio de la enfermedad. Se presenta disartria que evoluciona a anartria. Trastornos de la deglución, que llegan a requerir uso de gastrostomía para la nutrición, siendo un factor de mal pronóstico su aparición precoz. También se presentan otros síntomas y signos de liberación piramidal como la risa y llanto espasmódico, reflejo de Babinski, exaltación de reflejos osteotendíneos. La PSP se presenta habitualmente en la séptima década de la vida. Es un cuadro que progresa rápidamente con importante trastornos del equilibrio, inestabilidad de la marcha y caídas frecuentes, lo cual suele ser el primer síntoma. Durante el primer año de evolución, se asocian disartria y un síndrome acineto-rígido que no responde al tratamiento con levodopa. En la mitad de los casos durante el primer año de evolución aparecen los trastornos conductuales, sin embargo, es poco frecuente que éstos sean los primeros síntomas. Los trastornos de la mirada conjugada vertical, que son característicos de la enfermedad, están presentes durante el primer año en más de la mitad de los casos, pero en muchos de ellos pueden aparecer en estados más tardíos de la enfermedad. La sobrevida media desde el inicio de la enfermedad es de aproximadamente 5 a 7 años. Aquellos pacientes que presentan caídas frecuentes durante el primer año de evolución tienen una sobrevida más corta. La causa de muerte más frecuente son los cuadro broncopulmonares secundarios a aspiración. El diagnóstico diferencial con los otros síndromes acineto-rígidos, como la enfermedad de Parkinson, atrofia multisistémica, degeneración córtico basal, demencia de cuerpos de Lewy, enfermedad de Alzheimer, enfermedad cerebrovascular multinfarto, habitualmente es muy difícil, especialmente, en las etapas iniciales de estas enfermedades. Los criterios clínicos de diagnóstico no permiten certeza. Criterios clínicos para el diagnóstico de la PSP: Criterios mayores de inclusión Enfermedad gradualmente progresiva. Inicio después de los 40 años Trastorno de la mirada vertical de tipo supranuclear Inestabilidad postural, con caídas frecuentes de aparición precoz Criterios de apoyo • Síndrome acineto-rígido simétrico con rigidez de predominio proximal. • Postura de cuello anormal (retrocollis). • Pobre o ausente respuesta del parkinsonismo al uso de levodopa o agonistas dopaminérgicos. • Disfagia y disartria de inicio precoz. • Trastorno cognitivo de tipo frontal con inicio precoz.
 y también mutaciones polimerasa - gamma ( POLG ) heredadas en el modo dominante. Pacientes PSP- símil (parkinsonismo, parálisis mirada vertical y disfunción cognitiva precoz) alrededor de los 60 años. Sordera, ataxia y signos de neurona motora inferior. Ptosis y oftalmoplejía infranuclear, migraña y epilepsia. Excelente respuesta a la L- dopa. Steele, Richardson y Olszewski describieron esta enfermedad en 1964, también conocida como Parálisis Supranuclear Progresiva (PSP). Se caracteriza por trastornos de la mirada conjugada vertical, especialmente hacia abajo, parálisis pseudobulbar, disartria, distonia axial y demencia, Tb blefarospasm y retrocolis. Después de los cuarenta años y, en la mayoría de los casos, alrededor de los sesenta años. Trastornos motores que se presentan como un cuadro de parkinsonismo acinético rígido asociado a una pérdida de los reflejos posturales con caídas frecuentes que aparecen dentro del primer año de evolución, lo cual lleva a la imposibilidad de mantenerse en pie. Los pacientes presentan un aumento del tono muscular axial respecto al distal con extensión del cuello en retrocollis, adoptando una postura erecta en hiperextensión, que le da una apariencia de majestuosidad (distonia axial). El trastorno de la mirada se caracteriza por un enlentecimiento y, finalmente, la imposibilidad de ejecutar los movimientos oculares en especial en la mirada vertical conjugada hacia abajo. En el adulto mayor con frecuencia se pueden encontrar trastornos de la mirada conjugada hacia arriba y de la convergencia que no tienen significado patológico. El trastorno de la mirada conjugada observado en la PSP está presente en los movimientos voluntarios y de seguimiento, sin embargo, esta limitación es menor en los movimientos oculares reflejos, como al obtener el reflejo oculocefálico, en que se le gira activamente la cabeza al paciente y se obtiene el fenómeno conocido como ojos de muñecas . Con frecuencia este reflejo no es explorado en el plano vertical. Estos trastornos son de presentación gradual pudiendo llegar con la evolución de la enfermedad a un oftalmoplejía, en que se compromete la mirada conjugada en todos los sentidos e incluso en un pequeño número de pacientes se puede perder la respuesta refleja. En la primera etapa de la enfermedad se compromete la velocidad de los movimientos sacádicos de los ojos. En muchos casos se presenta una hipometría y descomposición de los movimientos sacádicos, similar al fenómeno conocido en la literatura anglosajona como square wave jerks . Se puede asociar a blefaroespasmo que, en algunos casos, se comporta como una apraxia parpebral, es decir, una dificultad para abrir voluntariamente los ojos por la inhibición supranuclear del músculo elevador de los párpados. Varias enfermedades neurológicas se presentan con trastornos de la mirada (adaptado de Lees A,1995): Atrofia Multisistémica tipo olivopontocerebelosa. Degeneración Corticobasal. Atrofia Dento-palido-nigro-lusial. Niemann-Pick de inicio tardío. Enfermedad de Joseph. Los trastornos conductuales y neuropsiquiátricos, más frecuentes son el síndrome frontal, se presentan como una falla de la función ejecutiva, apatía, cambio de personalidad, prensión forzada, perseveración de las conductas, enlentecimiento de pensamiento, disminución de la atención, disminución de la fluencia verbal. En muchos casos, se constituye un cuadro de demencia de tipo fronto-límbico. El síndrome pseudobulbar está presente, en todos los pacientes, en diferentes grados y puede ser uno de los síntomas de inicio de la enfermedad. Se presenta disartria que evoluciona a anartria. Trastornos de la deglución, que llegan a requerir uso de gastrostomía para la nutrición, siendo un factor de mal pronóstico su aparición precoz. También se presentan otros síntomas y signos de liberación piramidal como la risa y llanto espasmódico, reflejo de Babinski, exaltación de reflejos osteotendíneos. La PSP se presenta habitualmente en la séptima década de la vida. Es un cuadro que progresa rápidamente con importante trastornos del equilibrio, inestabilidad de la marcha y caídas frecuentes, lo cual suele ser el primer síntoma. Durante el primer año de evolución, se asocian disartria y un síndrome acineto-rígido que no responde al tratamiento con levodopa. En la mitad de los casos durante el primer año de evolución aparecen los trastornos conductuales, sin embargo, es poco frecuente que éstos sean los primeros síntomas. Los trastornos de la mirada conjugada vertical, que son característicos de la enfermedad, están presentes durante el primer año en más de la mitad de los casos, pero en muchos de ellos pueden aparecer en estados más tardíos de la enfermedad. La sobrevida media desde el inicio de la enfermedad es de aproximadamente 5 a 7 años. Aquellos pacientes que presentan caídas frecuentes durante el primer año de evolución tienen una sobrevida más corta. La causa de muerte más frecuente son los cuadro broncopulmonares secundarios a aspiración. El diagnóstico diferencial con los otros síndromes acineto-rígidos, como la enfermedad de Parkinson, atrofia multisistémica, degeneración córtico basal, demencia de cuerpos de Lewy, enfermedad de Alzheimer, enfermedad cerebrovascular multinfarto, habitualmente es muy difícil, especialmente, en las etapas iniciales de estas enfermedades. Los criterios clínicos de diagnóstico no permiten certeza. Criterios clínicos para el diagnóstico de la PSP: Criterios mayores de inclusión. Enfermedad gradualmente progresiva. Inicio después de los 40 años. Trastorno de la mirada vertical de tipo supranuclear. Inestabilidad postural, con caídas frecuentes de aparición precoz. Criterios de apoyo. • Síndrome acineto-rígido simétrico con rigidez de predominio proximal. • Postura de cuello anormal (retrocollis). • Pobre o ausente respuesta del parkinsonismo al uso de levodopa o agonistas dopaminérgicos. • Disfagia y disartria de inicio precoz. • Trastorno cognitivo de tipo frontal con inicio precoz.")
16
PSP-símil Priónicas Enfermedad de Creutzfeldt- Jakob Genética (mutaciones del gen de la proteína prionica PRNP) Inicio entre 5 y 7 década Parálisis mirada vertical, " apariencia facial preocupada " inestabilidad postural, rigidez axial y la demencia frontal (mutación E200K) Rapidez de la evolución, signos cerebelosos, espasticidad, y mioclonías sensible a estímulo RM hiperintensidades bilaterales en putamen y caudado en T2 y FLAIR, y DWI Proteína y EEG con patrones periódicos (puede ser normal inicialmente) Steele, Richardson y Olszewski describieron esta enfermedad en 1964, también conocida como Parálisis Supranuclear Progresiva (PSP). Se caracteriza por trastornos de la mirada conjugada vertical, especialmente hacia abajo, parálisis pseudobulbar, disartria, distonia axial y demencia, Tb blefarospasm y retrocolis. Después de los cuarenta años y, en la mayoría de los casos, alrededor de los sesenta años. Trastornos motores que se presentan como un cuadro de parkinsonismo acinético rígido asociado a una pérdida de los reflejos posturales con caídas frecuentes que aparecen dentro del primer año de evolución, lo cual lleva a la imposibilidad de mantenerse en pie. Los pacientes presentan un aumento del tono muscular axial respecto al distal con extensión del cuello en retrocollis, adoptando una postura erecta en hiperextensión, que le da una apariencia de majestuosidad (distonia axial). El trastorno de la mirada se caracteriza por un enlentecimiento y, finalmente, la imposibilidad de ejecutar los movimientos oculares en especial en la mirada vertical conjugada hacia abajo. En el adulto mayor con frecuencia se pueden encontrar trastornos de la mirada conjugada hacia arriba y de la convergencia que no tienen significado patológico. El trastorno de la mirada conjugada observado en la PSP está presente en los movimientos voluntarios y de seguimiento, sin embargo, esta limitación es menor en los movimientos oculares reflejos, como al obtener el reflejo oculocefálico, en que se le gira activamente la cabeza al paciente y se obtiene el fenómeno conocido como “ojos de muñecas”. Con frecuencia este reflejo no es explorado en el plano vertical. Estos trastornos son de presentación gradual pudiendo llegar con la evolución de la enfermedad a un oftalmoplejía, en que se compromete la mirada conjugada en todos los sentidos e incluso en un pequeño número de pacientes se puede perder la respuesta refleja. En la primera etapa de la enfermedad se compromete la velocidad de los movimientos sacádicos de los ojos. En muchos casos se presenta una hipometría y descomposición de los movimientos sacádicos, similar al fenómeno conocido en la literatura anglosajona como “square wave jerks”. Se puede asociar a blefaroespasmo que, en algunos casos, se comporta como una apraxia parpebral, es decir, una dificultad para abrir voluntariamente los ojos por la inhibición supranuclear del músculo elevador de los párpados. Varias enfermedades neurológicas se presentan con trastornos de la mirada (adaptado de Lees A,1995): Atrofia Multisistémica tipo olivopontocerebelosa. Degeneración Corticobasal. Atrofia Dento-palido-nigro-lusial. Niemann-Pick de inicio tardío. Enfermedad de Joseph. Los trastornos conductuales y neuropsiquiátricos, más frecuentes son el síndrome frontal, se presentan como una falla de la función ejecutiva, apatía, cambio de personalidad, prensión forzada, perseveración de las conductas, enlentecimiento de pensamiento, disminución de la atención, disminución de la fluencia verbal. En muchos casos, se constituye un cuadro de demencia de tipo fronto-límbico. El síndrome pseudobulbar está presente, en todos los pacientes, en diferentes grados y puede ser uno de los síntomas de inicio de la enfermedad. Se presenta disartria que evoluciona a anartria. Trastornos de la deglución, que llegan a requerir uso de gastrostomía para la nutrición, siendo un factor de mal pronóstico su aparición precoz. También se presentan otros síntomas y signos de liberación piramidal como la risa y llanto espasmódico, reflejo de Babinski, exaltación de reflejos osteotendíneos. La PSP se presenta habitualmente en la séptima década de la vida. Es un cuadro que progresa rápidamente con importante trastornos del equilibrio, inestabilidad de la marcha y caídas frecuentes, lo cual suele ser el primer síntoma. Durante el primer año de evolución, se asocian disartria y un síndrome acineto-rígido que no responde al tratamiento con levodopa. En la mitad de los casos durante el primer año de evolución aparecen los trastornos conductuales, sin embargo, es poco frecuente que éstos sean los primeros síntomas. Los trastornos de la mirada conjugada vertical, que son característicos de la enfermedad, están presentes durante el primer año en más de la mitad de los casos, pero en muchos de ellos pueden aparecer en estados más tardíos de la enfermedad. La sobrevida media desde el inicio de la enfermedad es de aproximadamente 5 a 7 años. Aquellos pacientes que presentan caídas frecuentes durante el primer año de evolución tienen una sobrevida más corta. La causa de muerte más frecuente son los cuadro broncopulmonares secundarios a aspiración. El diagnóstico diferencial con los otros síndromes acineto-rígidos, como la enfermedad de Parkinson, atrofia multisistémica, degeneración córtico basal, demencia de cuerpos de Lewy, enfermedad de Alzheimer, enfermedad cerebrovascular multinfarto, habitualmente es muy difícil, especialmente, en las etapas iniciales de estas enfermedades. Los criterios clínicos de diagnóstico no permiten certeza. Criterios clínicos para el diagnóstico de la PSP: Criterios mayores de inclusión Enfermedad gradualmente progresiva. Inicio después de los 40 años Trastorno de la mirada vertical de tipo supranuclear Inestabilidad postural, con caídas frecuentes de aparición precoz Criterios de apoyo • Síndrome acineto-rígido simétrico con rigidez de predominio proximal. • Postura de cuello anormal (retrocollis). • Pobre o ausente respuesta del parkinsonismo al uso de levodopa o agonistas dopaminérgicos. • Disfagia y disartria de inicio precoz. • Trastorno cognitivo de tipo frontal con inicio precoz.
 Rapidez de la evolución, signos cerebelosos, espasticidad, y mioclonías sensible a estímulo. RM hiperintensidades bilaterales en putamen y caudado en T2 y FLAIR, y DWI. Proteína y EEG con patrones periódicos (puede ser normal inicialmente) Steele, Richardson y Olszewski describieron esta enfermedad en 1964, también conocida como Parálisis Supranuclear Progresiva (PSP). Se caracteriza por trastornos de la mirada conjugada vertical, especialmente hacia abajo, parálisis pseudobulbar, disartria, distonia axial y demencia, Tb blefarospasm y retrocolis. Después de los cuarenta años y, en la mayoría de los casos, alrededor de los sesenta años. Trastornos motores que se presentan como un cuadro de parkinsonismo acinético rígido asociado a una pérdida de los reflejos posturales con caídas frecuentes que aparecen dentro del primer año de evolución, lo cual lleva a la imposibilidad de mantenerse en pie. Los pacientes presentan un aumento del tono muscular axial respecto al distal con extensión del cuello en retrocollis, adoptando una postura erecta en hiperextensión, que le da una apariencia de majestuosidad (distonia axial). El trastorno de la mirada se caracteriza por un enlentecimiento y, finalmente, la imposibilidad de ejecutar los movimientos oculares en especial en la mirada vertical conjugada hacia abajo. En el adulto mayor con frecuencia se pueden encontrar trastornos de la mirada conjugada hacia arriba y de la convergencia que no tienen significado patológico. El trastorno de la mirada conjugada observado en la PSP está presente en los movimientos voluntarios y de seguimiento, sin embargo, esta limitación es menor en los movimientos oculares reflejos, como al obtener el reflejo oculocefálico, en que se le gira activamente la cabeza al paciente y se obtiene el fenómeno conocido como ojos de muñecas . Con frecuencia este reflejo no es explorado en el plano vertical. Estos trastornos son de presentación gradual pudiendo llegar con la evolución de la enfermedad a un oftalmoplejía, en que se compromete la mirada conjugada en todos los sentidos e incluso en un pequeño número de pacientes se puede perder la respuesta refleja. En la primera etapa de la enfermedad se compromete la velocidad de los movimientos sacádicos de los ojos. En muchos casos se presenta una hipometría y descomposición de los movimientos sacádicos, similar al fenómeno conocido en la literatura anglosajona como square wave jerks . Se puede asociar a blefaroespasmo que, en algunos casos, se comporta como una apraxia parpebral, es decir, una dificultad para abrir voluntariamente los ojos por la inhibición supranuclear del músculo elevador de los párpados. Varias enfermedades neurológicas se presentan con trastornos de la mirada (adaptado de Lees A,1995): Atrofia Multisistémica tipo olivopontocerebelosa. Degeneración Corticobasal. Atrofia Dento-palido-nigro-lusial. Niemann-Pick de inicio tardío. Enfermedad de Joseph. Los trastornos conductuales y neuropsiquiátricos, más frecuentes son el síndrome frontal, se presentan como una falla de la función ejecutiva, apatía, cambio de personalidad, prensión forzada, perseveración de las conductas, enlentecimiento de pensamiento, disminución de la atención, disminución de la fluencia verbal. En muchos casos, se constituye un cuadro de demencia de tipo fronto-límbico. El síndrome pseudobulbar está presente, en todos los pacientes, en diferentes grados y puede ser uno de los síntomas de inicio de la enfermedad. Se presenta disartria que evoluciona a anartria. Trastornos de la deglución, que llegan a requerir uso de gastrostomía para la nutrición, siendo un factor de mal pronóstico su aparición precoz. También se presentan otros síntomas y signos de liberación piramidal como la risa y llanto espasmódico, reflejo de Babinski, exaltación de reflejos osteotendíneos. La PSP se presenta habitualmente en la séptima década de la vida. Es un cuadro que progresa rápidamente con importante trastornos del equilibrio, inestabilidad de la marcha y caídas frecuentes, lo cual suele ser el primer síntoma. Durante el primer año de evolución, se asocian disartria y un síndrome acineto-rígido que no responde al tratamiento con levodopa. En la mitad de los casos durante el primer año de evolución aparecen los trastornos conductuales, sin embargo, es poco frecuente que éstos sean los primeros síntomas. Los trastornos de la mirada conjugada vertical, que son característicos de la enfermedad, están presentes durante el primer año en más de la mitad de los casos, pero en muchos de ellos pueden aparecer en estados más tardíos de la enfermedad. La sobrevida media desde el inicio de la enfermedad es de aproximadamente 5 a 7 años. Aquellos pacientes que presentan caídas frecuentes durante el primer año de evolución tienen una sobrevida más corta. La causa de muerte más frecuente son los cuadro broncopulmonares secundarios a aspiración. El diagnóstico diferencial con los otros síndromes acineto-rígidos, como la enfermedad de Parkinson, atrofia multisistémica, degeneración córtico basal, demencia de cuerpos de Lewy, enfermedad de Alzheimer, enfermedad cerebrovascular multinfarto, habitualmente es muy difícil, especialmente, en las etapas iniciales de estas enfermedades. Los criterios clínicos de diagnóstico no permiten certeza. Criterios clínicos para el diagnóstico de la PSP: Criterios mayores de inclusión. Enfermedad gradualmente progresiva. Inicio después de los 40 años. Trastorno de la mirada vertical de tipo supranuclear. Inestabilidad postural, con caídas frecuentes de aparición precoz. Criterios de apoyo. • Síndrome acineto-rígido simétrico con rigidez de predominio proximal. • Postura de cuello anormal (retrocollis). • Pobre o ausente respuesta del parkinsonismo al uso de levodopa o agonistas dopaminérgicos. • Disfagia y disartria de inicio precoz. • Trastorno cognitivo de tipo frontal con inicio precoz.")
18
DCB-símil Genéticas Neurodegenerativas Neurometabólicas Priónicas DFT
Alzheimer Neurometabólicas Xantmatosis Cerebrotendinea Enfermedad de Gaucher Priónicas

19
DCB-símil Neurodegenerativas
DFT (mutaciones PGRN y C9ORF72) PGRN: DCB fenotipo frecuente y afasia puede preceder síntomas RM atrofia cortical asimétrica en ambos, incluyendo lóbulo parietal. C9ORF72: parkinsonismo con características variables parecidas a DCB. La presencia de células del asta anterior características y alucinaciones Mutaciones Fusionado-en-sarcoma (FUS) pueden causar síndromes de superposición ELA-parkinsonismo
 PGRN: DCB fenotipo frecuente y afasia puede preceder síntomas. RM atrofia cortical asimétrica en ambos, incluyendo lóbulo parietal. C9ORF72: parkinsonismo con características variables parecidas a DCB. La presencia de células del asta anterior características y alucinaciones. Mutaciones Fusionado-en-sarcoma (FUS) pueden causar síndromes de superposición ELA-parkinsonismo.")
20
DCB-símil Neurodegenerativas
Alzheimer (mutaciones Preseniles PSEN-1y APP) Síntomas DCB en pacientes con EA esporádica El aumento de la latencia sacádicos en ambas, parkinsonismo con mioclonías, distonía, apraxia y la demencia frontal Edad anterior en el inicio y mioclonías es más sugerente de EA, no hay asimetría tan marcada Pueden haber convulsiones, raro en DCB vez se producen en CBD
 Síntomas DCB en pacientes con EA esporádica. El aumento de la latencia sacádicos en ambas, parkinsonismo con mioclonías, distonía, apraxia y la demencia frontal. Edad anterior en el inicio y mioclonías es más sugerente de EA, no hay asimetría tan marcada. Pueden haber convulsiones, raro en DCB vez se producen en CBD.")
21
DCB-símil Neurometabólicas
Xantomatosis Cerebrotendinea (mutación CYP27A1) Raro error innato del metabolismo causado por mutaciones en el gen CYP27A1 Acumulación de colestanol , un compuesto tóxico , en el cerebro y otros tejidos Edad media de aparición 40 años Parkinsonismo (20%) asimétrico, apraxia, y distonía Síntomas cognitivos (93%), signos piramidales (93%), signos cerebelosos (53 %) y el 27% había caídas Mejora con L- dopa fue frecuente ( 91 %) pero modesto efecto Signos sistémicos importantes: catarata juvenil ( 93 %), los xantomas tendinosos (87%), la osteopenia y la aterosclerosis temprana. El engrosamiento de la cola de caballo RM cerebral muestra atrofia cerebelosa (100 %) y cambios señal núcleos dentados ( 80 %) DaTSCAN es anormal en xantomatosis cerebrotendinosa y en la mayoría DCB Diagnóstico: colestanol plasmática elevada y pruebas genéticas Tratamiento con ácido quenodesoxicólico y los inhibidores: inhibidores de la HMG - CoA reductasa y la 3 - hidroxi metil - glutaril - coenzima A. Normaliza hallazgos bioquímicos, mejora características clínicas y retrasa la progresión (contradictorio) RM tobillo
 Raro error innato del metabolismo causado por mutaciones en el gen CYP27A1. Acumulación de colestanol , un compuesto tóxico , en el cerebro y otros tejidos. Edad media de aparición 40 años. Parkinsonismo (20%) asimétrico, apraxia, y distonía. Síntomas cognitivos (93%), signos piramidales (93%), signos cerebelosos (53 %) y el 27% había caídas. Mejora con L- dopa fue frecuente ( 91 %) pero modesto efecto. Signos sistémicos importantes: catarata juvenil ( 93 %), los xantomas tendinosos (87%), la osteopenia y la aterosclerosis temprana. El engrosamiento de la cola de caballo. RM cerebral muestra atrofia cerebelosa (100 %) y cambios señal núcleos dentados ( 80 %) DaTSCAN es anormal en xantomatosis cerebrotendinosa y en la mayoría DCB. Diagnóstico: colestanol plasmática elevada y pruebas genéticas. Tratamiento con ácido quenodesoxicólico y los inhibidores: inhibidores de la HMG - CoA reductasa y la 3 - hidroxi metil - glutaril - coenzima A. Normaliza hallazgos bioquímicos, mejora características clínicas y retrasa la progresión (contradictorio) RM tobillo.")
22
DCB-símil Neurometabólicas
Enfermedad de Gaucher Casos DCB simil descrito: síndrome asimétrica, acinético rígido, distonía de extremidades, apraxia y levitación de extremidades, con cambios cognitivos y de comportamiento con ledad de inicio de 60 años También como demencia, parkinsonismo, mioclonías, lentitud de movimientos sacádicos horizontales, pero sin apraxia extremidades.

23
DCB-símil Priónicas Priónicas
DCB-símil en 1 familia irlandesa con CJDg con gran variabilidad fenotípica que se encontró que tenía una mutación en el codón 117 del gen PRNP

25
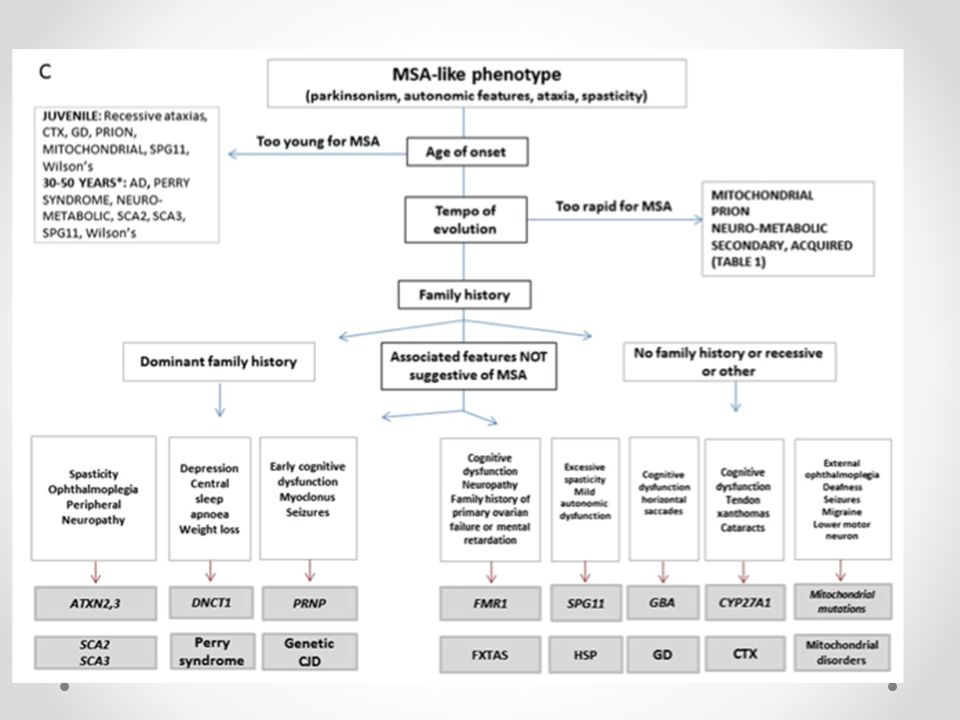
AMS-símil Genéticas Neurodegenerativas Neurometabólicas Priónicas
Síndrome X-Frágil Ataxias Espinocerebelosas Neurometabólicas Priónicas

26
AMS-símil Neurodegenerativas
Síndrome X-Frágil (Retraso Mental Gene1 con premutación carriers con X Frágil) Síndrome de temblor - ataxia X frágil ( FXTAS ) es un inicio tardío ( > 50 años de edad ) premutación CGG con expansión ( 55 a 200 repeticiones) La penetrancia de FXTAS en portadoras de sexo masculino mayores de 50 años es del 40% Mujeres portadoras con poca frecuencia desarrollan FXTAS . El diagnóstico puede realizarse mediante pruebas genéticas La autopsia revela inclusiones intranucleares en las neuronas y los astrocitos y la materia blanca distrófica . El fenotipo típico consiste en la combinación de temblor de intención y ataxia , con parkinsonismo , disfunción autonómica , deterioro cognitivo , características psiquiátricos y neuropatía periférica Una historia familiar de retraso mental o insuficiencia ovárica prematura RM mayor intensidad de la señal en los pedúnculos cerebelosos. El borde putaminal característica se describe en el MSA no se ha descrito en FXTAS DaTSCAN en FTXAS puede ser normal o anormal
 Síndrome de temblor - ataxia X frágil ( FXTAS ) es un inicio tardío ( > 50 años de edad ) premutación CGG con expansión ( 55 a 200 repeticiones) La penetrancia de FXTAS en portadoras de sexo masculino mayores de 50 años es del 40% Mujeres portadoras con poca frecuencia desarrollan FXTAS . El diagnóstico puede realizarse mediante pruebas genéticas. La autopsia revela inclusiones intranucleares en las neuronas y los astrocitos y la materia blanca distrófica . El fenotipo típico consiste en la combinación de temblor de intención y ataxia , con parkinsonismo , disfunción autonómica , deterioro cognitivo , características psiquiátricos y neuropatía periférica. Una historia familiar de retraso mental o insuficiencia ovárica prematura. RM mayor intensidad de la señal en los pedúnculos cerebelosos. El borde putaminal característica se describe en el MSA no se ha descrito en FXTAS. DaTSCAN en FTXAS puede ser normal o anormal.")
27
AMS-símil Neurodegenerativas
Ataxias Espinocerebelosas SCA3 (enfermedad de Machado -Joseph ) es la causa más frecuente de herencia autosómica dominante heredado ataxia cerebelosa La edad de inicio varía entre 5 y 75 años , e inversamente correlacionada con la longitud de repetición CAG Ataxia cerebelosa , parkinsonismo y sólo la disfunción cognitiva leve, puede haber disfunción autonómica estar presente Oftalmoplejía ( 56 %) y neuropatía periférica RM contracción moderada del vermis cerebeloso y hemisferios y atrofia pontina en ambas DaTSCAN es anormal en ambas SCA6 con parkinsonismo, con fenotipo más leve y sin disfunción autonómica
 es la causa más frecuente de herencia autosómica dominante heredado ataxia cerebelosa. La edad de inicio varía entre 5 y 75 años , e inversamente correlacionada con la longitud de repetición CAG. Ataxia cerebelosa , parkinsonismo y sólo la disfunción cognitiva leve, puede haber disfunción autonómica estar presente. Oftalmoplejía ( 56 %) y neuropatía periférica. RM contracción moderada del vermis cerebeloso y hemisferios y atrofia pontina en ambas. DaTSCAN es anormal en ambas. SCA6 con parkinsonismo, con fenotipo más leve y sin disfunción autonómica.")
29
Aproximación Diagnóstica
Enfoque de diagnóstico de 4 pasos a un paciente que se presenta con " atípica " parkinsonismo atípico. Edad de inicio es crucial En segundo lugar, el ritmo de progresión El tercer paso sería una historia familiar detallada Carácterísticas atípicas

31
Después de haber reducido los posibles diagnósticos diferenciales por edad de inicio, ritmo de progresión , y la historia familiar , el cuarto paso crucial es buscar las características clínicas asociadas que apuntarán hacia el diagnóstico mutaciones particulares . Examen neurológico cuidadoso es importante para recoger las señales que son "atípicas " para PSP , CBD , y MSA , y dictará los posibles diagnósticos alternativos. Las características clínicas de la PSP , CBD , y MSA están bien descritos , ya pesar de los diferentes fenotipos se han descrito recientemente para estos trastornos , todavía hay algunas señales de que nunca están presentes , por ejemplo la ataxia , a diferencia de la inestabilidad postural, no es de el espectro fenotípico de PSP y el CDB , así un fenotipo PSP o CBD , con ataxia adicional, debe plantear la sospecha de otros trastornos ; o un paciente que se presenta con un aspecto similar imagen de PSP , pero con PEC horizontal prominente y temprana podría tener SCA2 o la enfermedad de Gaucher . Del mismo modo, temprano y prominente disfunción cognitiva o espasticidad severa sería muy inusual en MSA , y deben hacer sospechar de otros trastornos . Por otra parte , las características sistémicas son útiles cuando se sospecha un trastorno neurometabólica , e incluso pueden determinar el diagnóstico , como en xantomatosis cerebrotendinosa , cuando xantomas tendinosos están presentes. Se necesita esta detallada caracterización fenotípica antes de investigaciones específicas o se solicita la prueba genética . Obviamente , este algoritmo es una guía clínica y debe ser seguido más o menos , y no en términos absolutos y no cubre todo el espectro fenotípico de parkinsonismo atípico en trastornos genéticos. Sin embargo , dado que la lista de enfermedades genéticas que causan parkinsonismo atípico está en constante aumento , guías fenotípicas tales como que hemos presentado son importantes para reducir el número de posibles diagnósticos y , investigaciones innecesarias , piezas que requieren mucho tiempo , y de imagen caro moleculares y genéticos. conclusión Aquí hemos considerado trastornos genéticos que pueden cursar con un fenotipo similar a PSP , CBD -como, o MSA -como y hemos proporcionado pistas sobre que características ayudarán a los médicos a sospechar un trastorno genético subyacente en estos pacientes. La lista de enfermedades genéticas que causan PSP , CBD , y MSA parecidos es cada vez mayor ; Por lo tanto , los algoritmos de diagnóstico son importantes para ayudar a identificar correctamente a estos pacientes . El correcto diagnóstico diferencial es importante para el pronóstico y el tratamiento, y para la investigación futura en estos trastornos.

Presentaciones similares



















 Novedades>")


