Descargar la presentación
La descarga está en progreso. Por favor, espere

1
Errores Innatos Del Metabolismo Y Sindrome Parkinsoniano
Francisca Montoya Salvadores Residente de Neurología Universidad de los Andes

2
Errores innatos del metabolismo
Término acuñado por Archibald Garrod, 1927 Hasta hace poco, limitado a pediatría: RN e infancia Trastornos monogénicos producen la alteración de una enzima Trastorno en la síntesis o catabolismo de proteínas carbohidratos o lípidos Disrupción de las vías metabólicas Acumulación tóxica de sustratos, metabolitos intermedios de vías alternativas y/o defectos en la producción y/o utilización de energía Indiidualmente raros, colectivamente numerosos

3
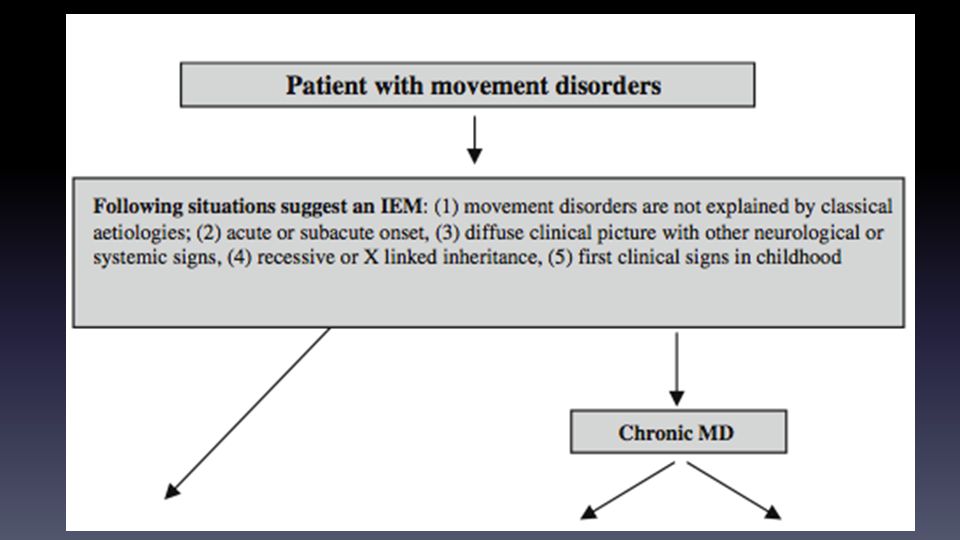
Clasificaciones Compromiso de cualquier órgano o sistema
Perfil temporal: agudo-grave vs. subagudo, progresivo degenerativo Todos pueden aparecer en adultos CLASIFICACIÓN CLÁSICA Trastornos en el metabolismo proteico Trastornos en el metabolismo de carbohidratos Trastornos en el almacenamiento lisosomal Defecto en la oxidación de ácidos grasos Trastornos mitocondriales Trastornos peroxisomales

4
Presentación psiquiátrica o neurológica
Inicio adultez ( >70 ) Afecta organelos: lisosomas, peroxisomas, defectos de síntesis de colesterol y glicosilación Presentación psiquiátrica o neurológica Psicosis atípica, depresión, coma, NP periférica, ataxia cerebelosa, paraparesia espástica, demencia, epilepsia y trastornos del movimiento Interfiere con la embriogénesis: dismorfias, displasia y malformaciones Adultos con encefalopatía: Intoxicación: RM N: Ciclo de la urea, porfirias, defectos de metilación de homocisteína Metabolismo energético: RM alt: Trastorno de cadena respiratoria, deficiencia de piruvato deshidrogenasa, biotina No interfiere con el desarrollo embrionario: intervalo libre Ataque metabólico agudo Crónico Todos los 3 grupos pueden presentarse en adultos, pero los tipo I son los mas frecuentes Tipo 3: GB muy vulnerables a trastornos energéticos y metales
 Afecta organelos: lisosomas, peroxisomas, defectos de síntesis de colesterol y glicosilación. Presentación psiquiátrica o neurológica. Psicosis atípica, depresión, coma, NP periférica, ataxia cerebelosa, paraparesia espástica, demencia, epilepsia y trastornos del movimiento. Interfiere con la embriogénesis: dismorfias, displasia y malformaciones. Adultos con encefalopatía: Intoxicación: RM N: Ciclo de la urea, porfirias, defectos de metilación de homocisteína. Metabolismo energético: RM alt: Trastorno de cadena respiratoria, deficiencia de piruvato deshidrogenasa, biotina. No interfiere con el desarrollo embrionario: intervalo libre. Ataque metabólico agudo. Crónico. Todos los 3 grupos pueden presentarse en adultos, pero los tipo I son los mas frecuentes. Tipo 3: GB muy vulnerables a trastornos energéticos y metales.")
5
Errores innatos del metabolismo y trastornos del movimiento
Se pueden presentar en la adolescencia o adultez Trastornos del movimiento asociados: PKS, distonía, corea, tics, mioclonus Causas determinadas por la sensibilidad de los GB a: Enfermedades por depósito de metales Trastornos del metabolismo energético Trastornos por depósito lisosomal Defectos en la síntesis de NT*

8
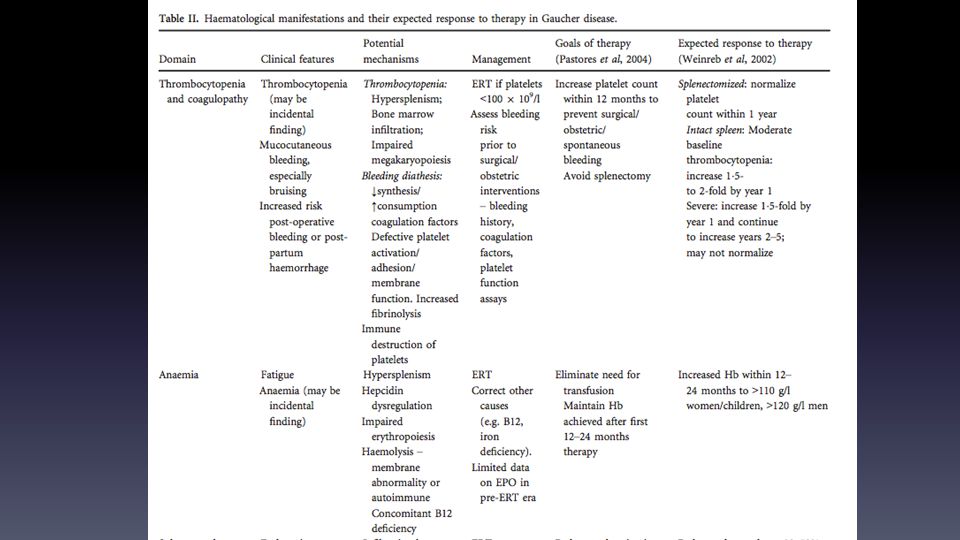
Enfermedad de Gaucher Trastorno lisosomal AR: deficiencia de la glucocerebrosidasa Síntomas Neurológicos: Epilepsia mioclónica progresiva, PKS, oftalmoplejia horizontal supranuclear, ataxia, Otros síntomas: Astenia, hepatoesplenomegalia, trombopenia, anemia, manifestaciones óseas Exámenes: de la actividad de la glucocerebrosidasa: en leu periféricos o cultivos de fibroblastos en piel biopsiada Células de Gaucher en bx MO: MQ grandes, cargados de lípidos con un nu excéntrico e inclusiones fibrilares citoplasmáticas levemente basófilas Genético: Gen Glucocerebrosidasa: cromosoma 1q21

9
Enfermedad de Gaucher Tipo I: 95% gaucher en caucásicos

10
Metabolismo de los glicoesfingolípidos

11
Fisiopatología Estado heterocigoto de mutación GBA en PKS y DCL: rol patógeno más allá de la deficiencia enzimática Ubicuitina-Proteosoma

12
Tratamientos Tx - esplenectomía y cx ortopédica Trasplante alogénico MO (1980) Terapia de reemplazo hormonal 3 productos con eficacia en parámetros hematológicos y vicerales Terapia de reducción de sustratos Miglustat: Inhibe la síntesis de glucosilceramide Única que atraviesa la BHE Chaperonas Mutaciones patogénicas en el gen GBA llevan a mal plegamiento proteico y degradación prematura Chaperonas se unen a las proteínas en el retículo endoplásmico, las estabilizan y ayudan a mantener la correcta conformación

15
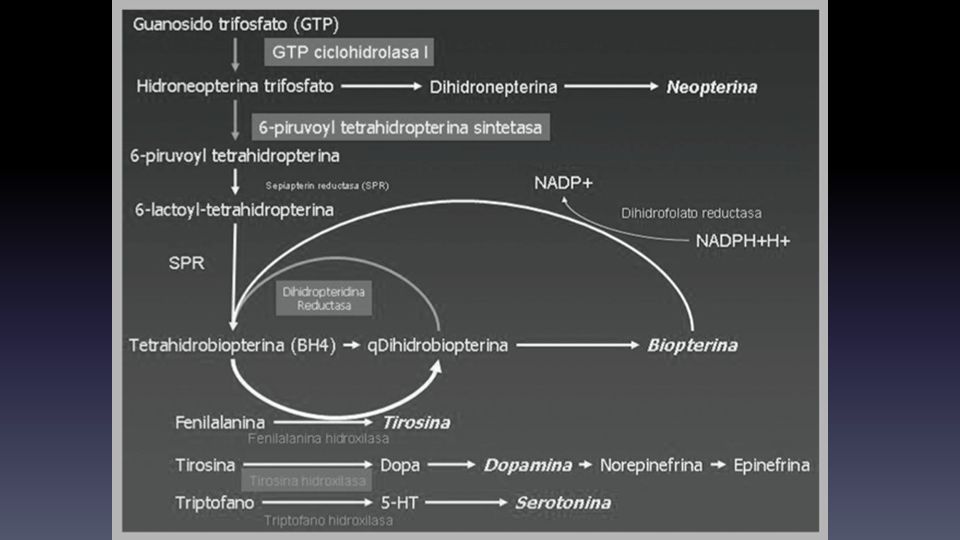
Defectos en la síntesis de las monoaminas
Defectos en las vías de biosíntesis de aminas biogénicas Estudio: medición directa de NT en LCR Salvo la deficiencia de la GTP ciclohidrolasa I Hiperprolactinemia indica defecto en la síntesis de dopamina Ejemplos Enfermedad de Segawa: deficiencia de la ciclohidrolasa I, AD mutación gen GTPCH1 Se presenta a cualquier edad con distonía o PKS, paraparesia pseudoespástica, fluctuaciones diurnas Exámenes: biopterinas, neopterinas HVA y 5-HIAA en LCR N o . Test de carga de Fenilalanina anormal Tratamiento: Levodopa, anticolinesterásicos, agonistas dopaminérgicos

16
Defectos en la síntesis de las monoaminas
Ejemplos Deficiencia de tirosina hidrolasa, AR Síntomas: distonía o PKS con poca o nula respuesta a L-dopa, paraparesia pseudoespástica, signos piramidales y RM Exámenes: HVA en LCR con biopterinas, neopterinas y 5HIAA Ns Tratamiento: Levodopa, anticolinesterásicos, agonistas dopaminérgicos Deficiencia de dihidropteridina reductasa, AR Síntomas: RM, distonia, PKS, epilepsia, depresión Exámenes: hiperfenilalaninemia, biopterinas, HVA y 5HIAA en LCR Tratamiento: Levodopa, 5HTP, dieta baja en fenilalanina

18
Hiperfenilalaninemia
Trastorno intermediario del metabolismo, AR Síntomas neurológicos PKS, atrofia óptica, demencia, leucoencefalopatía Exámenes: hiperfenilalaninemia, hipotirosinemia Tratamiento: Dieta baja en fenilalanina

19
Xantomatosis cerebrotendínea
Trastorno del metabolismo intermediario AR Síntomas neurológicos: Ataxia cerebelosa, PKS, paraparesia espástica, trastornos psiquiátricos Otros síntomas: Cataratas juveniles, xantomas tendíneos, diarrea RM: hiperintensidad en nucleo dentado Exámenes: colestanol elevado Tratamiento: Ácido quenodeoxycólico

21
Hemocromatosis Enfermedad de depósito AR – mut C282Y
Síntomas Neurológicos: PKS, ataxia cerebelosa, demencia, temblor de intención, mioclonias, distonía, sd piramidal Síntomas Sistémicos: Trastornos hepáticos, artritis, cardiomiopatía, coloración bronceada de la piel, astenia RM: N o atrofia cerebral Exámenes: Fe sérico alto, saturación de la transferrina y feritina. Tratamiento: Flebotomía (no eficiente en PKS) PKS puede o no responder a L-dopa,
 PKS puede o no responder a L-dopa,")
22
Trastornos de la cadena respiratoria
Trastorno del metabolismo energético Cualquier tipo de herencia: buscar herencia mitocondrial o mutaciones del DNA nuclear específicas Síntomas Neurológicos PKS, distonía, mioclonias Otros síntomas dependen del sd (oftalmoplejia externa, PNP, endocrinopatía, retinitis pigmentosa) Exámenes: aumento del lactato en LCR Bx: fibras rojas rasgadas Tratamiento: sintomático
 Exámenes: aumento del lactato en LCR. Bx: fibras rojas rasgadas. Tratamiento: sintomático.")
23
Lipofuscinosis ceroidea neuronal
Trastorno neurodegenerativo progresivo en niños secundario a acumulación de lipopigmentos autofluorescentes en las neuronas y glias Herencia AR Clínica: Epi mioclónica progresiva asociada a trastornos visuales 4 formas: Infantil (1,5 ±0,5á), infantil tardío (3,5 ±0,5á), Juvenil (6,5 1,8±á) Adulto (44 ±05,6) Regresión de los hitos del DSM, convulsiones, mioclonias, corea, amaurosis y ataxia Adultos: Trastorno de comportamiento y signos extrapiramidales Muerte 5 a á en las formas infantiles y a los 15-25á en las juveniles. Adulto tiene curso variable
, infantil tardío (3,5 ±0,5á), Juvenil (6,5 1,8±á) Adulto (44 ±05,6) Regresión de los hitos del DSM, convulsiones, mioclonias, corea, amaurosis y ataxia. Adultos: Trastorno de comportamiento y signos extrapiramidales. Muerte 5 a á en las formas infantiles y a los 15-25á en las juveniles. Adulto tiene curso variable.")
24
Lipofuscinosis ceroidea neuronal
Exámenes EEG: enlentecimiento difuso y generalizado, con descargas epilépticas en 81% Otros: SSEP gigantes, VEP P100 prolongados, EMG: neuropatía axonal; atrofia difusa en neuroimágenes Bx: Cerebro, piel músculo hígado Cerebro: atrofia del manto cortical, deplesión neuronal y presencia de astrocios reactivos. Tinción PAS y luxol fast-blue revelan inclusiones intracitoplasmáticas intensas, granulares. Microscopio luz fluorescente: sustancia amarilla autofluorescente Bx piel y músculo son Ns en microscopá de luz. Estudios ultraestructurales muestran las inclusiones

25
Niemann-Pick C disease
Trastorno lisosomal, AR Se presenta a cualquier edad: depósito de esfingomielina Clínica: mioclonias, distonía, corea, PKS, trastornos psiquiátricos, parálisis supranuclear de la mirada vertical, ataxia cerebelosa, demencia Otros: hepatoesplenomegalia

26
Deficiencia primaria de la actividad ácida de la esfingomielinasa (A y B)
Deficiencia del procesamiento celular y del transporte de colesterol LDL

27
Niemann-Pick C disease
Exámenes: tinción filipina anormal en fibroblastos Se reduce la esterificación y se produce una excesiva acumulación de colesterol MRI: N o atrofia cerebelosa Tratamiento: Miglustat: inhibidor de la biosíntesis de glicoesfingolípidos Reduce el depósito de lípidos, mejora la captación endosómicay normaliza el tráfico de lípidos en los linfocitos B Datos limitados de que su uso puedan prevenir o reducir la progresión de la enfermedad

28
Enfermedad de Wilson Trastorno metabolismo de metales, AR, mutación del gen ATP7b: transportador Cu-ATPasa – Acumulación de Cu en cerebro (GB) e hígado Ceruloplasmina y ferritina juegan un rol crítico en el metabolismo de metales en el cerebro Ceruloplasmina: actividad ferroxidasa para liberar el Fe de su sitio de almacenaje Ferritina: fuente de Fe intracelular Cu es un cofactor que interviene en la movilización del Fe Cu se fija a la apoceruloplasmina, le da estabilidad y evita su degradación. Clínica: Adultos jóvenes 7ma década Temblor, PKS, distonía, corea, signos psiquiátricos, disartria
 e hígado. Ceruloplasmina y ferritina juegan un rol crítico en el metabolismo de metales en el cerebro. Ceruloplasmina: actividad ferroxidasa para liberar el Fe de su sitio de almacenaje. Ferritina: fuente de Fe intracelular. Cu es un cofactor que interviene en la movilización del Fe. Cu se fija a la apoceruloplasmina, le da estabilidad y evita su degradación. Clínica: Adultos jóvenes 7ma década. Temblor, PKS, distonía, corea, signos psiquiátricos, disartria.")
29
Enfermedad de Wilson Otros: anillos de kayser-Fleischer, DHC
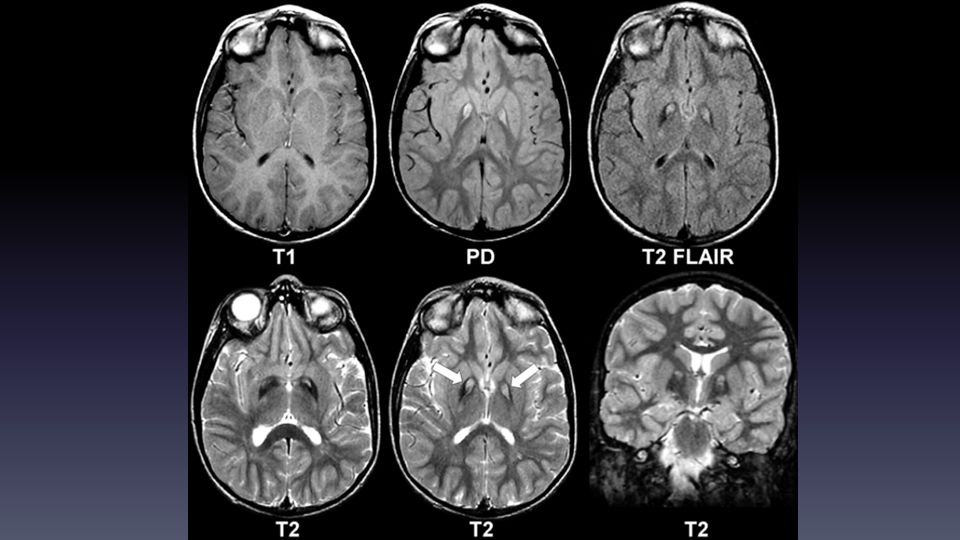
Exámenes: Cu urinario , Cu sérico y ceruloplasmina RM: depósitos de metales en GB Atrofia cerebral T1 en nu lentiforme y mesencéfalo en estadio inicial T2 en globo pálido T2 caudado, putamen, tálamo VL , nu dentado y puente Tratamiento: D-penicilamina, zinc, trientina

31
Neurodegeneración asociada a la pantotenato kinasa: Hallervorden-Spatz
Trastorno metabólico metales, AR, mutación gen PANK2 Pank2 cataliza la síntesis de coenzima A desde vitamina B5 Presenta desde la primera década Clínica: PKS, distonía, disartria, trastornos psiquiátricos y cognitivos, signos piramidales, acantocitosis (8-10%) retinitis pigmentosa RM: signo de los ojos de tigre T2 en el globo pálido con un central T2 GRE: á de susceptibilidad magnética Manejo: sintomático
 retinitis pigmentosa. RM: signo de los ojos de tigre. T2 en el globo pálido con un central. T2 GRE: á de susceptibilidad magnética. Manejo: sintomático.")
33
Conclusión Trastornos metabólicos son poco frecuentes de forma aislada
Algunos se pueden presentar con trastornos del movimiento: diagnóstico diferencial parkinsonismos Alto nivel de sospecha Importancia: algunas son tratables y definir pronóstico

Presentaciones similares








>")











. Forma primaria: herencia autosómica recesiva Alteración.>")
 y electrones (citocromos)>")


 ¿ENFERMEDADES RARAS?>")