Descargar la presentación
La descarga está en progreso. Por favor, espere

1
SISTEMA BILIAR DRA. MONIKA NUÑEZ

2
Patología de Sistema biliar
Anomalía congénitas Ausencia congénita de la V.B Duplicación congénita Vesícula bilobulada V.B con fondo doblado es la anomalía más frecuente Localización aberrante de la vesícula biliar Se da en 5-10% de la población Vesícula parcial o completamente metida en el parénquima hepático Agenesia de parte o toda la vía biliar común o de conductos hepáticos Atresia de la vía biliar extrahepática

3
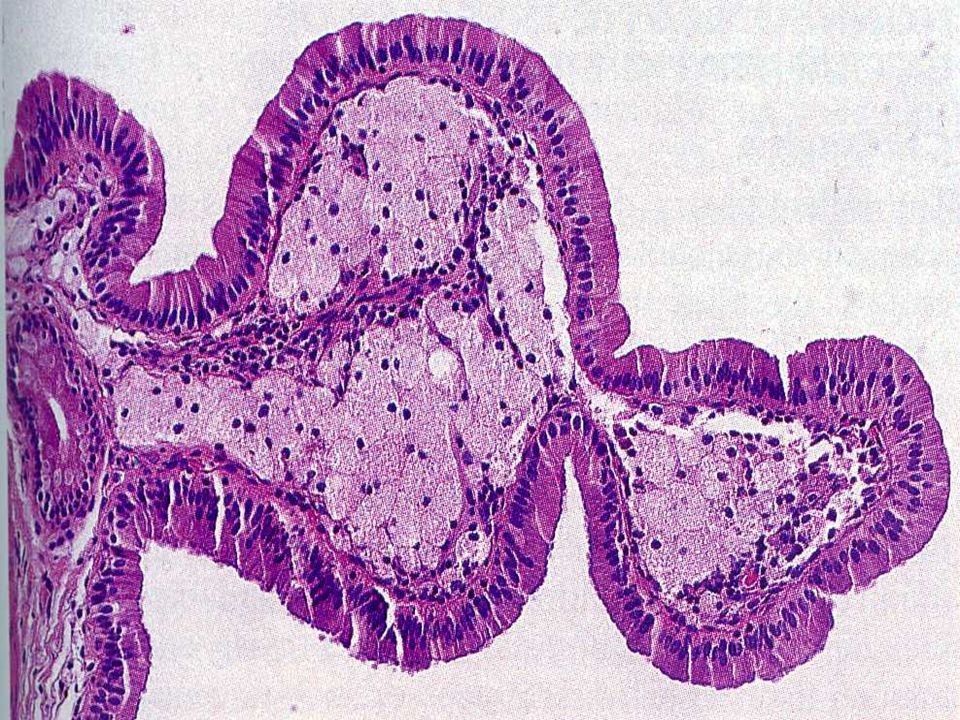
Vesícula Biliar cuyo fondo está plegado hacia adentro Vesícula biliar En Gorro de Frigio Dolor abdominal en niños y adulto joven

5
V.B septada clínica es dolor
abdominal en niños y adulto joven

7
Patología de la vía biliar
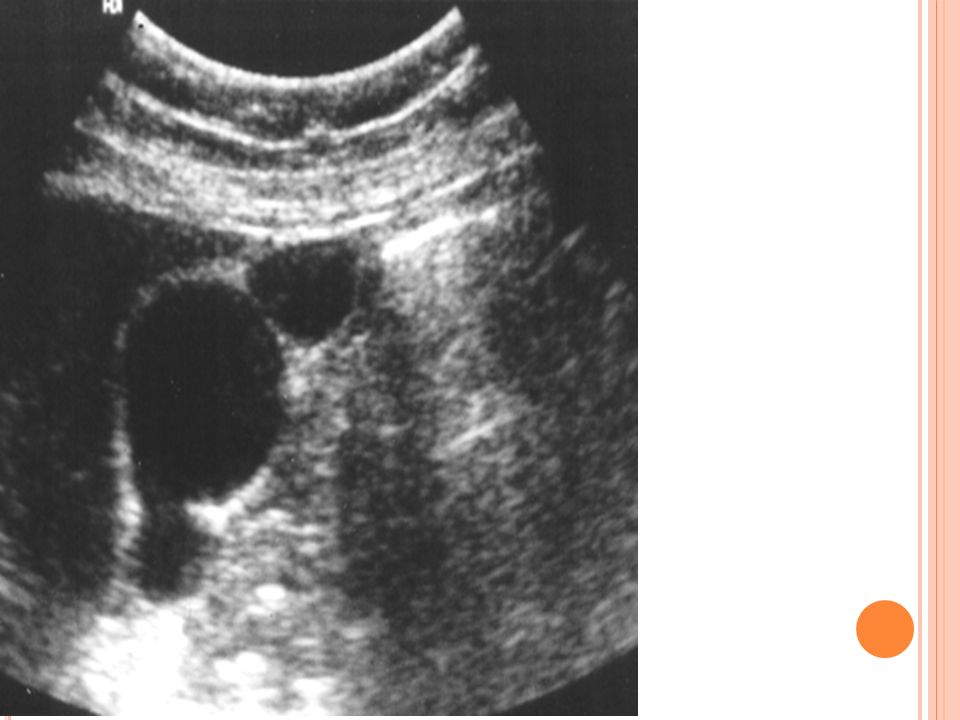
Los trastornos de la vía biliar afectan a una importante proporción de la población mundial Más del 95% de la patología de la vía biliar está dada por piedras en la vesícula: COLELITIASIS La patología más frecuente de la vesícula biliar es la colelitiasis

8
Colelitiasis 10-20% población adulta en países desarrollados
Un millón de casos nuevos/año van a colecistectomía cada año Occidente 90% de litiasis son por colesterol: cálculos amarillos El otro 10% son litos por sales de calcio: cálculos pigmentados

9
Clínica de Colelitiasis
Dolor tipo cólico espasmódico en hipocondrio derecho Sin embargo más de 80% son silentes o asintomáticas por décadas

11
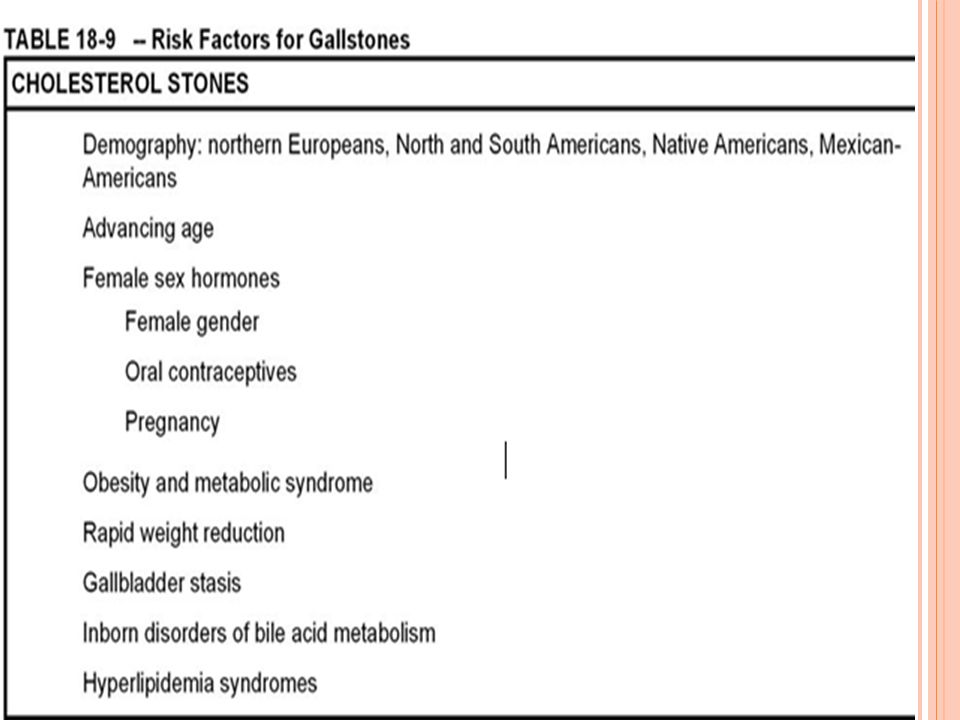
Colelitiasis: prevalencia y factores de riesgo
Etnia y geografía: 75% nativos americanos. Ej. Navajos Sociedades industrializadas Edad y sexo: 5% población antes de 40 años 30% mayores de 40 años 2 veces más en mujeres que hombres Factores ambientales: Influencia estrogénica: anticonceptivos, embarazo Obesidad

13
Colelitiasis: prevalencia y factores de riesgo
Factores hereditarios: Síndromes de hiperlipidemias con marcado incremento de biosíntesis de colesterol Otros factores de riesgo: Síndromes hemolíticos Disfunción ileal severa Infecciones bacterianas del árbol biliar

14
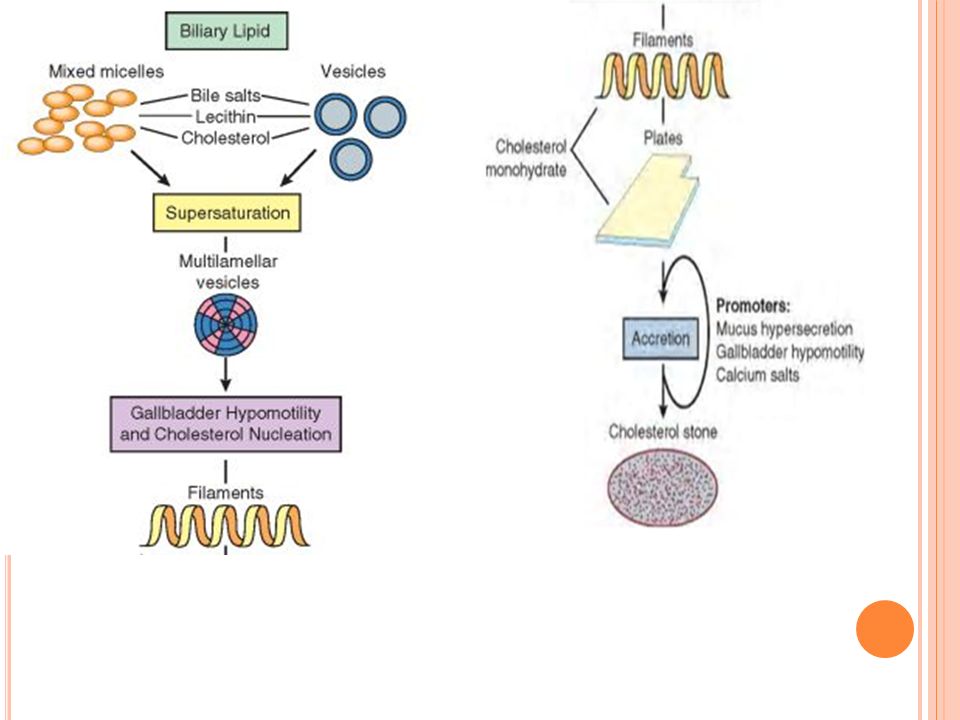
Patogénesis de cálculos colesterol
Para que se formen las piedras de colesterol se requiere: Bilis sobresaturada con colesterol (concentraciones de colesterol exceden la capacidad de solubilidad de la bilis dada por el efecto detergente de sus sales biliares y lecitinas) Colesterol ya no está disperso, se agrega formando núcleos sólidos de cristales de monohidrato de colesterol Hipomotilidad de la vesícula biliar promueve la nucleación Nucleación del colesterol en la bilis se ve acelerada Hipersecreción de moco en la V.B atrapa los cristales nucleados y promueve la agregación de los mismos para formar cálculos
 Colesterol ya no está disperso, se agrega formando núcleos sólidos de cristales de monohidrato de colesterol. Hipomotilidad de la vesícula biliar promueve la nucleación. Nucleación del colesterol en la bilis se ve acelerada. Hipersecreción de moco en la V.B atrapa los cristales nucleados y promueve la agregación de los mismos para formar cálculos.")
17
Patogénesis de cálculos pigmentados
Son una mezcla compleja de sales de calcio insolubles, bilirrubina no conjugada y sales de calcio inorgánicas Causas Niveles altos de bilirrubina no conjugada en la bilis Síndromes hemolíticos Disfunción ileal Contaminación bacteriana del árbol biliar Escherichia coli Ascaris lumbricoides Opisthorchis sinensis (Asia) 8% de cálculos de colesterol y cálculos pigmentados no se identifica factor de riesgo asociado
 8% de cálculos de colesterol y cálculos pigmentados no se identifica factor de riesgo asociado.")
19
Colecistitis aguda Se define como un episodio de dolor biliar agudo con dolor en hipocondrio derecho, con persistencia de síntomas por más de 24 horas 90% casos se asocia a colelitiasis >mujeres de años Dolor en cuadrante superior derecho, signo de Murphy (dolor al palpar VB en inspiración profunda)
")
20
Colecistitis aguda calculosa
Oclusión del cuello de V.B o del cístico por el lito Causa dilatación y edema de la pared Ya a las 48h hay bacterias como E coli, enterococos y anaerobios colonizando la V.B Histología: pared engrosada y edematosa, congestiva y hemorrágica, con abundantes neutrófilos

21
Colecistitis aguda calculosa
Presentación clínica: Paciente febril Puede tener ictericia Leucocitosis Complicaciones: Empiema Necrosis de V.B Perforación Peritonitis biliar y séptica

22
Colecistitis aguda acalculosa
5% de colecistectomías Factores de riesgo: Trauma Cirugía abdominal mayor no biliar Sepsis Quemaduras severas Nutrición parenteral prolongada Múltiples transfusiones sanguíneas Uso de narcóticos Deshidratación Anestesia Estado post parto Mortalidad de 5-10%

23
Colecistitis aguda enfisematosa
Colecistitis aguda con producción de gas por infección bacteriana (clostridium) y aterosclerosis de la arteria cística. 50-60 años Diabéticos Mortalidad 15%
 y aterosclerosis de la arteria cística años. Diabéticos. Mortalidad 15%")
24
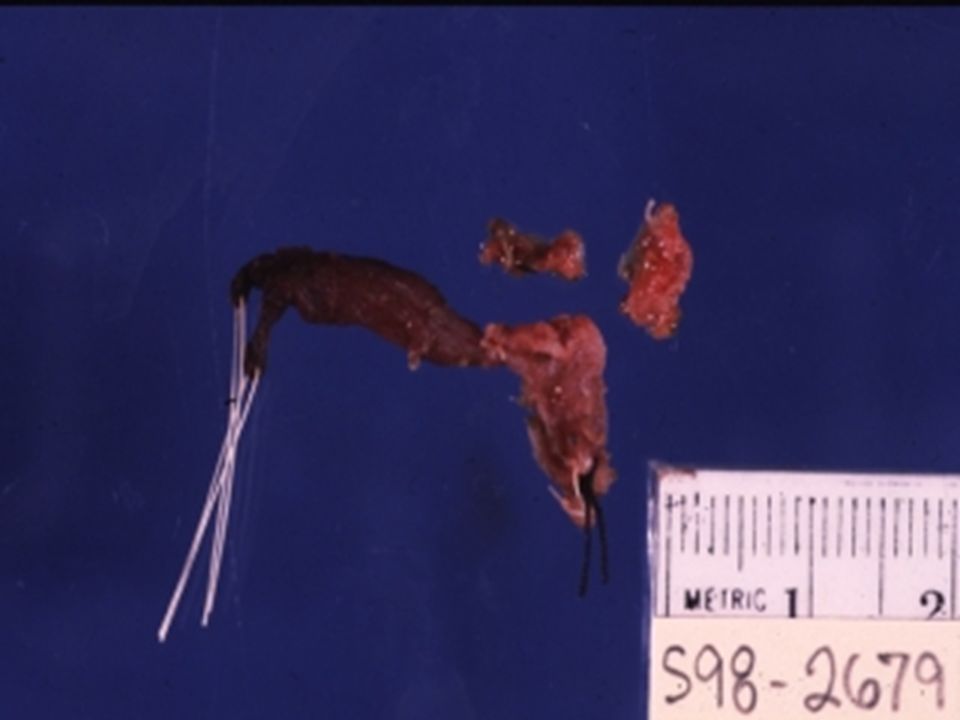
PERFORACION COLECISTITIS AGUDA

26
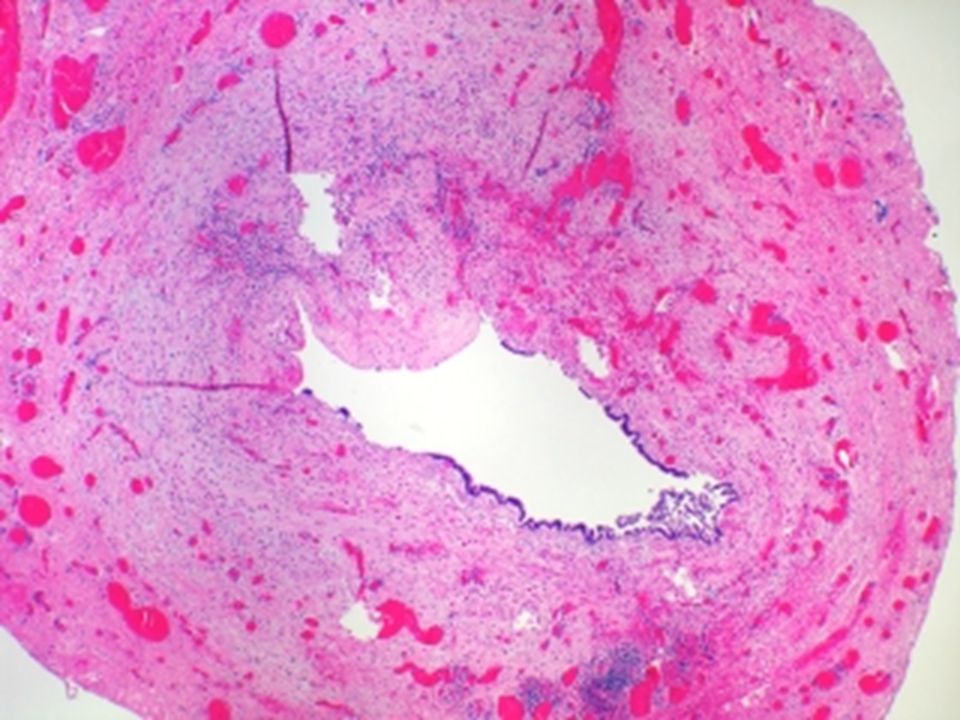
Colecistitis crónica Se asocia a colelitiasis
Son secundarias a colecistitis agudas leve a repetición Histología: inflamación mononuclear, predominio de linfocitos y fibrosis de la pared

27
COLECISTITIS CRÓNICA

29
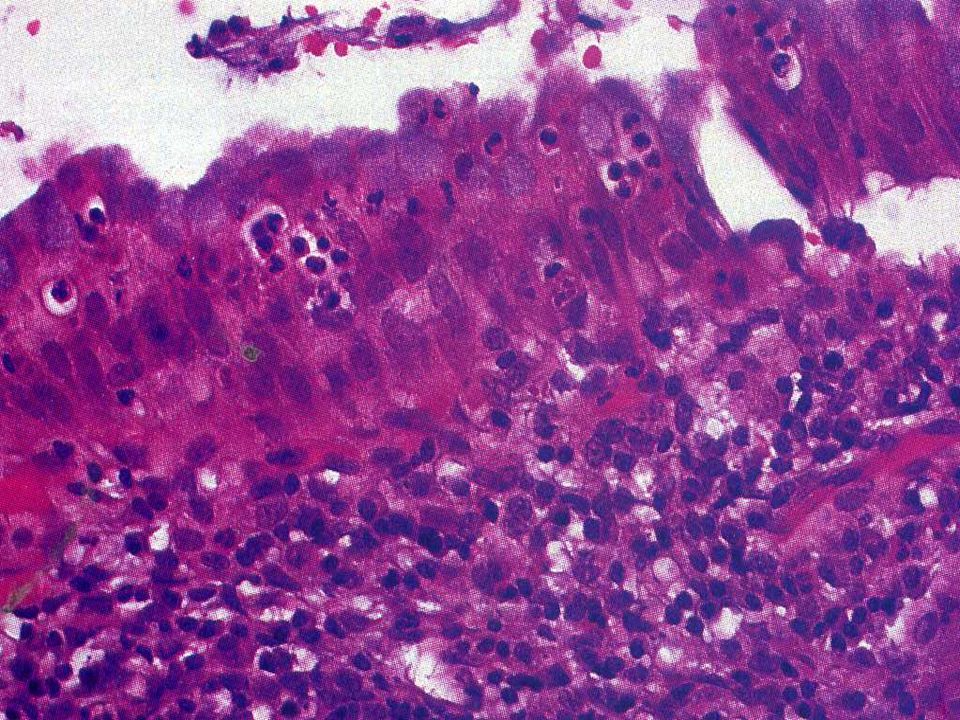
Colesterolosis Presencia de macrófagos con lípidos en su citoplasma a nivel de la lámina propia

31
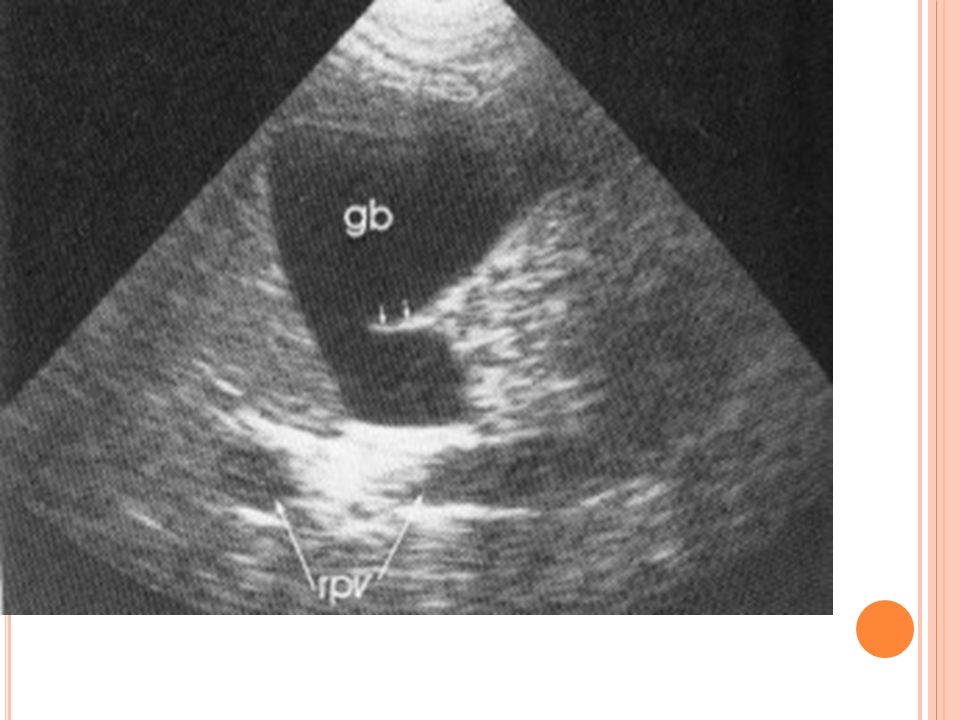
Hidrops de Vesícula biliar
Vesícula mucoide clara Piedra impactada en cuello de V.B En niños puede confundirse con adenitis mesentérica

32
Colangitis ascendente
Va de la mano con la coledocolitiasis Piedra en árbol biliar 10% de colelitiasis O por formación de novo de cálculos en las vías biliares luego de colecistectomía Asia: mayor incidencia en formación de litiasis en árbol biliar

33
Causas de colangitis ascendente
Obstrucción vía biliar Pancreatitis Colangitis Absceso hepático Cirrosis biliar secundaria Colecistitis aguda acalculosa

34
Colangitis ascendente
Colangitis significa infección bacteriana de los conductos biliares Resulta de la obstrucción del flujo, principalmente por litiasis Otras causas: catéteres, tumores, pancreatitis aguda

35
Colangitis ascendente
Bacterias Aerobios Gram negativos: E. coli Klebsiella Clostridium Bacteroides E. Enterobacter Estreptococo del grupo D

36
Clínica de colangitis ascendente
Fiebre Escalofríos Dolor abdominal Ictericia Histología: infiltrado inflamatorio dado por neutrófilos en lumen de conductos Puede llevar a absceso hepático

37
Desordenes de conductos extrahepáticos (Atresia biliar)
Atresia biliar: 1/3 de colestasis neonatal = 1/ nacidos vivos Se caracteriza por obstrucción completa del flujo biliar Neonatos con colestasis Bilirrubina total 6-12mg/dl Elevación de transaminasa y fosfatasa alcalina Tratamiento inicial es by pass del árbol biliar Progreso rápido a cirrosis biliar Requiere trasplante de hígado a temprana edad

38
Atresia biliar extra hepática Patogénesis
Son niños que nacen con árbol biliar sano. Luego inicia infiltrado inflamatorio del árbol biliar con fibrosis y obliteración de la luz La atresia biliar se define como la obstrucción parcial o completa del lumen del árbol biliar extrahepático que sucede en los primero tres meses de vida Causa desconocida, se ha propuesto lo siguiente: Infección viral: retrovirus tipo 3, citomegalovirus, rubéola virus Factor genético Desarrollo embrionario anómalo: 20% con anomalías extra hepáticas y atresia biliar Son pacientes con poliesplenismo, defecto cardiovascular, malrotación, atresia intestinal

39
Atresia de conductos intrahepáticos 6% Atresia de conducto biliar común 3% Atresia de vesícula, cístico y conducto hepático común. 19% Atresia extra hepática completa 72%

42
hepatoportojejunostomía

43
Colangitis esclerosante primaria
> hombres hasta 45 años 50% con enfermedad inflamatoria intestinal tipo CUCI O CROHN, tiroiditis, sarcoidosis, histiocitosis x, artritis reumatoide, etc Enfermedad progresiva Mortalidad es de 6 años después del inicio de síntomas Fatiga, prurito, ictericia y lento desarrollo a cirrosis

44
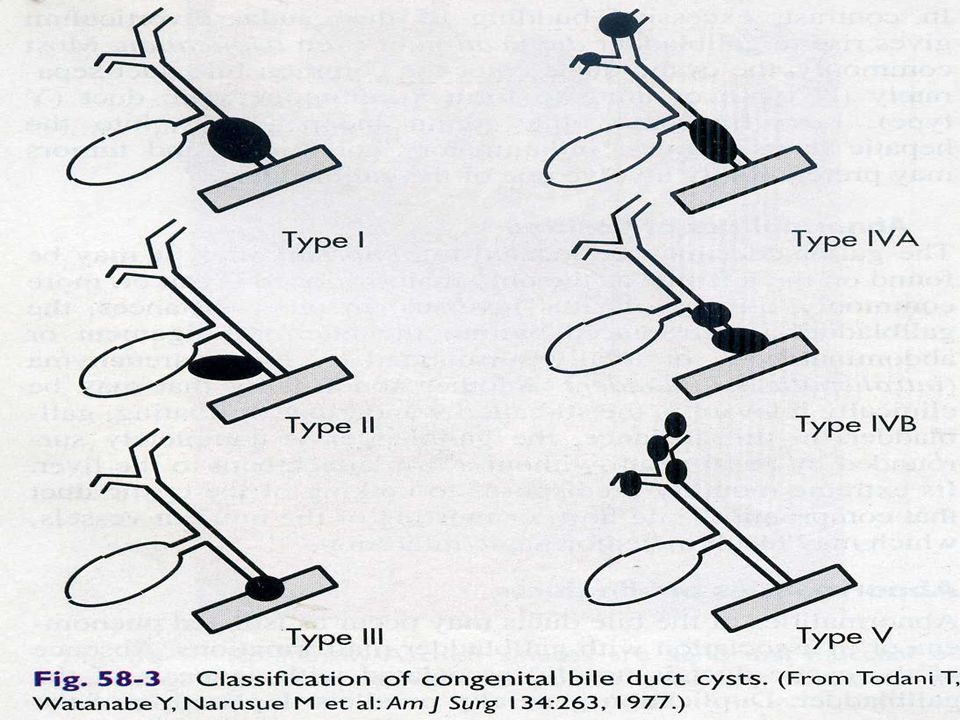
Quiste de colédoco Dilatación congénita de conducto biliar común
Niños menores de 10 años con ictericia y dolor abdominal tipo cólico 20% inicia síntomas en adultez Mujer 1:4 hombres Síndrome de Caroli se asocia a dilatación quística de la vía biliar intra hepática

46
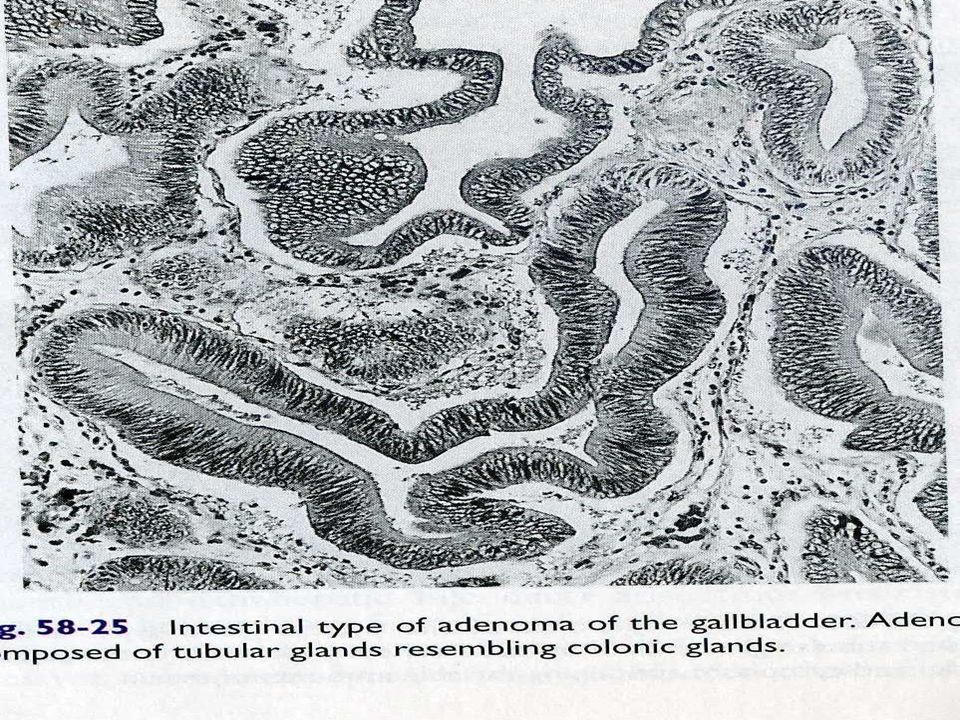
Neoplasias Pólipos de V.B
Adenoma Tubular Papilar Túbulo-papilar

50
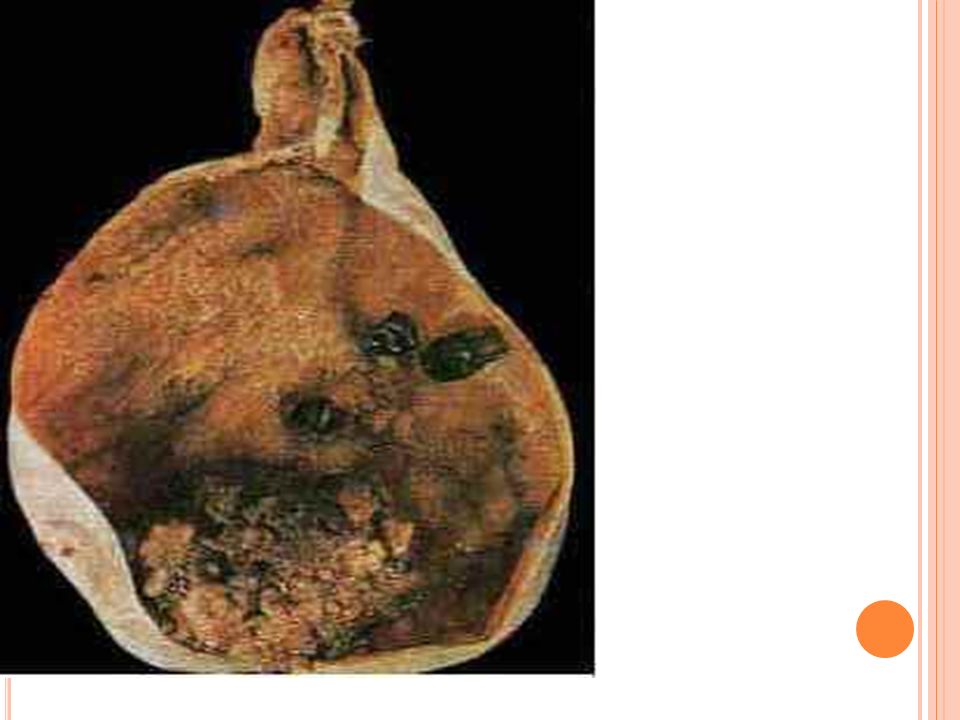
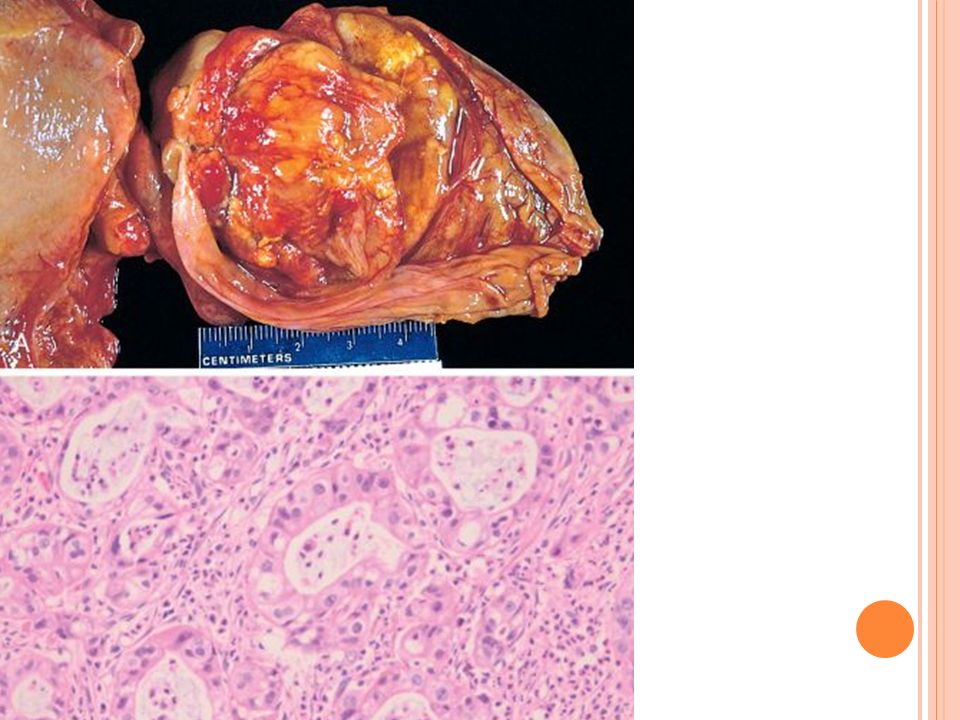
Carcinoma de la vesícula biliar
Quinto cáncer más común del SGI en USA Pico de años Pico para carcinoma de la vía biliar años Si es invasor es irresecable 1% sobrevida a 5 años Se asocia a colelitiasis en 90% en occidente Asia: 100% es infecciosa o parasitaria, especialmente por Opistorchis sinensis

51
Carcinoma de VB Puede ser plano o fúngico Localización: fondo y cuello
Morfología Ultrasonido Puede ser plano o fúngico Localización: fondo y cuello Casi siempre adenocarcinomas 5% son carcinomas escamoso o tienen diferenciación adeno escamosa

54
Páncreas

55
PANCREATITIS AGUDA Muchas complicaciones ocurren en los pacientes con grado inicial D o E, presentando una tasa de mortalidad del 14% y de morbilidad de 54%.

56
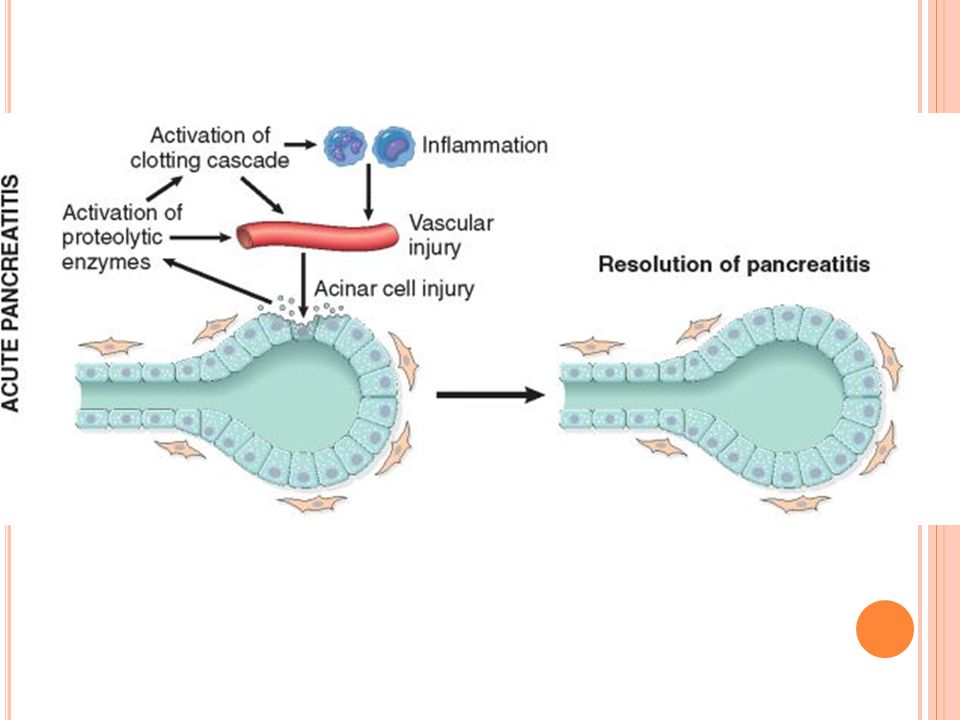
PANCREATITIS AGUDA Inflamación aguda de una glándula pancreática previamente sana, por una inadecuada activación intracelular de las enzimas pancreáticas

59
PANCREATITIS AGUDA Etiología Litiasis biliar 30-75% Alcoholismo
Mujeres > 60a Alcoholismo Hombres Idiopática (20%) .....Microlitiasis??? Niños traumatismos enfermedades sistémicas 85-95% Muchas complicaciones ocurren en los pacientes con grado inicial D o E, presentando una tasa de mortalidad del 14% y de morbilidad de 54%.
 .....Microlitiasis Niños traumatismos. enfermedades sistémicas % Muchas complicaciones ocurren en los pacientes con grado inicial D o E, presentando una tasa de mortalidad del 14% y de morbilidad de 54%.")
60
PANCREATITIS AGUDA Etiología Hipertrigliceridemia
Post-ERCP o post-IQ (estómago, via biliar…) Obstrucción del conducto pancreático ( tumores, páncreas divisum o anular) Fármacos ( azatioprina, clortiazida, estrógenos,furosemida, sulfamidas, tetraciclinas, penicilina….) Infecciones (hepatitis, parotiditis, rubeola, CMV, cándida, ascaris, Schistosoma) Hiperparatiroidismo Muchas complicaciones ocurren en los pacientes con grado inicial D o E, presentando una tasa de mortalidad del 14% y de morbilidad de 54%.
 Obstrucción del conducto pancreático ( tumores, páncreas divisum o anular) Fármacos ( azatioprina, clortiazida, estrógenos,furosemida, sulfamidas, tetraciclinas, penicilina….) Infecciones (hepatitis, parotiditis, rubeola, CMV, cándida, ascaris, Schistosoma) Hiperparatiroidismo. Muchas complicaciones ocurren en los pacientes con grado inicial D o E, presentando una tasa de mortalidad del 14% y de morbilidad de 54%.")
61
PANCREATITIS AGUDA Fisiopatología NORMAL PANCREATITIS AGUDA
Enzimas duodenales Enzimas pancreáticos inactivos (pro-enzimas) Pro-enzimas pasan al espacio extracelular y duodeno Enzimas activos PANCREATITIS AGUDA Muchas complicaciones ocurren en los pacientes con grado inicial D o E, presentando una tasa de mortalidad del 14% y de morbilidad de 54%. Litiasis-Alcohol Alteración barrera protección celular?? Enzimas duodenales Enzimas pancreáticos inactivos (pro-enzimas) Activación intraglandular e intracelular Autodigestión (necrosis)
 Pro-enzimas pasan al espacio extracelular y duodeno. Enzimas activos. PANCREATITIS AGUDA. Muchas complicaciones ocurren en los pacientes con grado inicial D o E, presentando una tasa de mortalidad del 14% y de morbilidad de 54%. Litiasis-Alcohol Alteración barrera protección celular Enzimas duodenales. Enzimas pancreáticos. inactivos. (pro-enzimas) Activación intraglandular e intracelular. Autodigestión. (necrosis)")
63
PANCREATITIS AGUDA Diagnóstico clínico Dolor (85-100%)
-Inicio súbito (ingesta o abuso alcohol) epigástrico, irradiado a ambos hipocondrios y a la espalda, difuso. Náuseas y vómitos (54-92%) Distensión abdominal (paresia intestinal) Exploración física: Fiebre, taquicardia, taquipnea, derrame pleural, ictericia, hipotensión,shock, equimosi (signos de Cullen y Grey Turner) Muchas complicaciones ocurren en los pacientes con grado inicial D o E, presentando una tasa de mortalidad del 14% y de morbilidad de 54%.
 epigástrico, irradiado a ambos hipocondrios y a la espalda, difuso. Náuseas y vómitos (54-92%) Distensión abdominal (paresia intestinal) Exploración física: Fiebre, taquicardia, taquipnea, derrame pleural, ictericia, hipotensión,shock, equimosi (signos de Cullen y Grey Turner) Muchas complicaciones ocurren en los pacientes con grado inicial D o E, presentando una tasa de mortalidad del 14% y de morbilidad de 54%.")
64
PANCREATITIS AGUDA Equimosis (signos de Cullen y Grey Turner <1%).
Muchas complicaciones ocurren en los pacientes con grado inicial D o E, presentando una tasa de mortalidad del 14% y de morbilidad de 54%.

65
PANCREATITIS AGUDA Diagnóstico bioquímico Amilasa
Leucocitosis Elevación GPT, Bil, FA, GGT, GOT (origen biliar) Disminución del calcio (formas necrohemorrágicas) Elevación niveles séricos de enzimas pancreáticos : Amilasa >3 veces el valor normal Muy sensible, poco específico. Lipasa Más específico, dura 3-5 días Muchas complicaciones ocurren en los pacientes con grado inicial D o E, presentando una tasa de mortalidad del 14% y de morbilidad de 54%.
 Disminución del calcio (formas necrohemorrágicas) Elevación niveles séricos de enzimas pancreáticos : Amilasa. >3 veces el valor normal. Muy sensible, poco específico. Lipasa. Más específico, dura 3-5 días. Muchas complicaciones ocurren en los pacientes con grado inicial D o E, presentando una tasa de mortalidad del 14% y de morbilidad de 54%.")
66
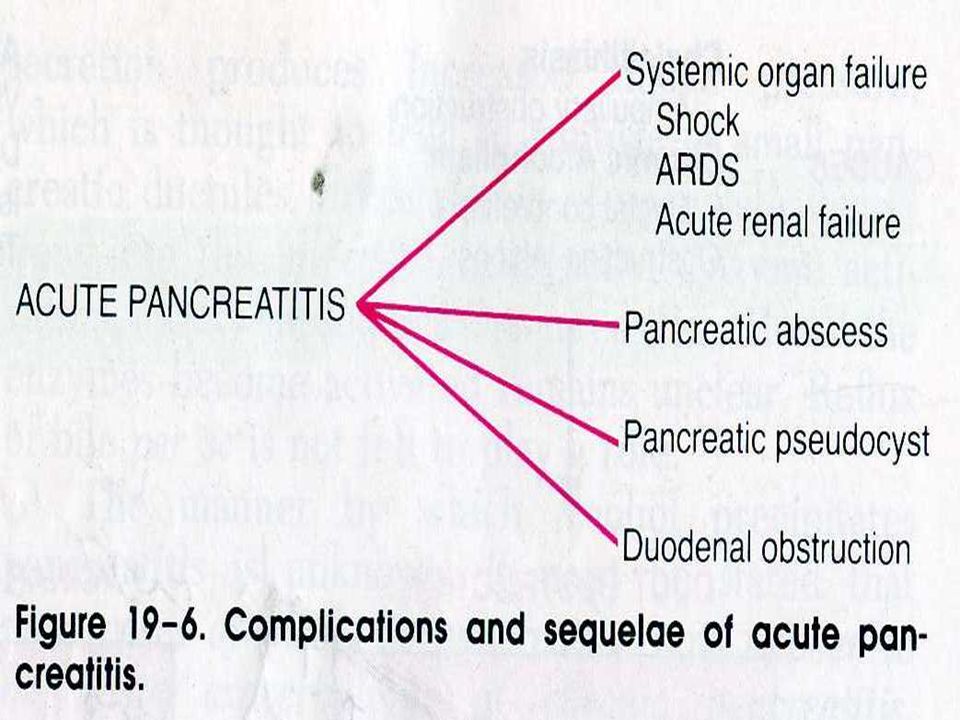
PANCREATITIS AGUDA Manifestaciones morfológicas
Páncreas normal o de tamaño. Inflamación peripancreática. Complicaciones locales Líquido intraabdominal Necrosis pancreática PRECOCES Muchas complicaciones ocurren en los pacientes con grado inicial D o E, presentando una tasa de mortalidad del 14% y de morbilidad de 54%. Absceso Pseudoquiste Complicaciones vasculares TARDÍAS >4 semanas

67
PANCREATITIS AGUDA Absceso Colección de pus circunscrita
TC: colección líquida con paredes +/-gruesas que captan contraste ev CON o SIN gas en su interior. > 4 semanas Muchas complicaciones ocurren en los pacientes con grado inicial D o E, presentando una tasa de mortalidad del 14% y de morbilidad de 54%.

68
PANCREATITIS AGUDA Pseudoquiste 50 % resolución espontánea
Colección líquida encapsulada Pseudo-pared no epitelializada (tejido de granulación / fibrosis) TC: colección líquida con pared que capta contraste ev > 4 semanas Muchas complicaciones ocurren en los pacientes con grado inicial D o E, presentando una tasa de mortalidad del 14% y de morbilidad de 54%. 50 % resolución espontánea 50 % estabilización o complicación
 TC: colección líquida con pared que capta contraste ev. > 4 semanas. Muchas complicaciones ocurren en los pacientes con grado inicial D o E, presentando una tasa de mortalidad del 14% y de morbilidad de 54%. 50 % resolución espontánea. 50 % estabilización o complicación.")
69
PANCREATITIS AGUDA Pseudoquiste Complicaciones
Infección - Absceso (gas/sin gas) Vasculares - Sangrado - Formación pseudoaneurisma - Trombosis venosa Obstrucción intestinal / biliar Dolor abdominal Muchas complicaciones ocurren en los pacientes con grado inicial D o E, presentando una tasa de mortalidad del 14% y de morbilidad de 54%.
 Vasculares - Sangrado. - Formación pseudoaneurisma. - Trombosis venosa. Obstrucción intestinal / biliar. Dolor abdominal. Muchas complicaciones ocurren en los pacientes con grado inicial D o E, presentando una tasa de mortalidad del 14% y de morbilidad de 54%.")
72
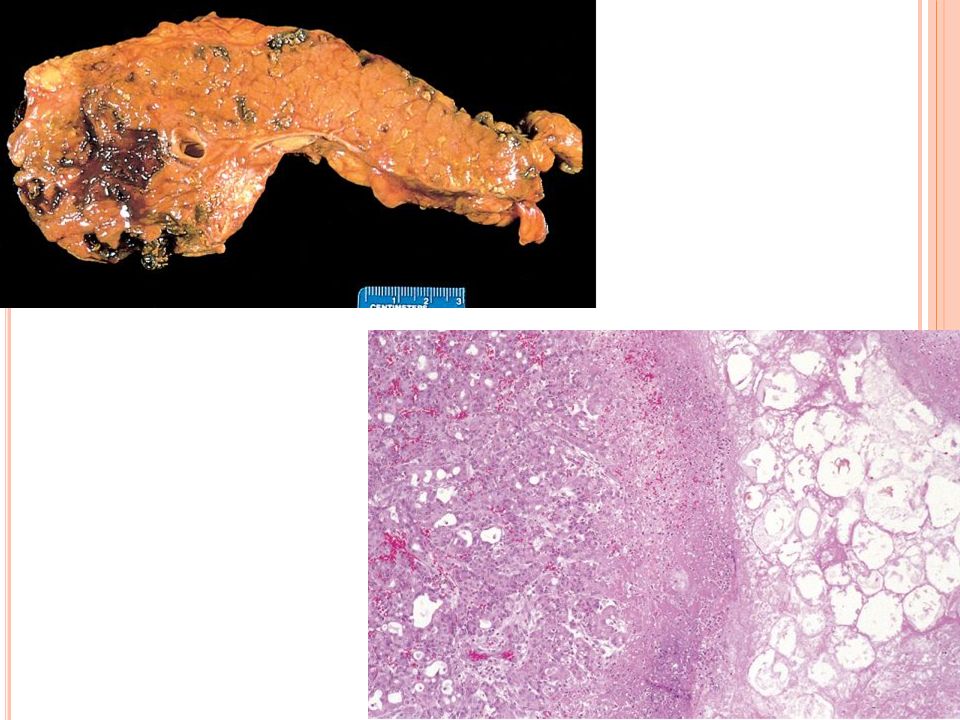
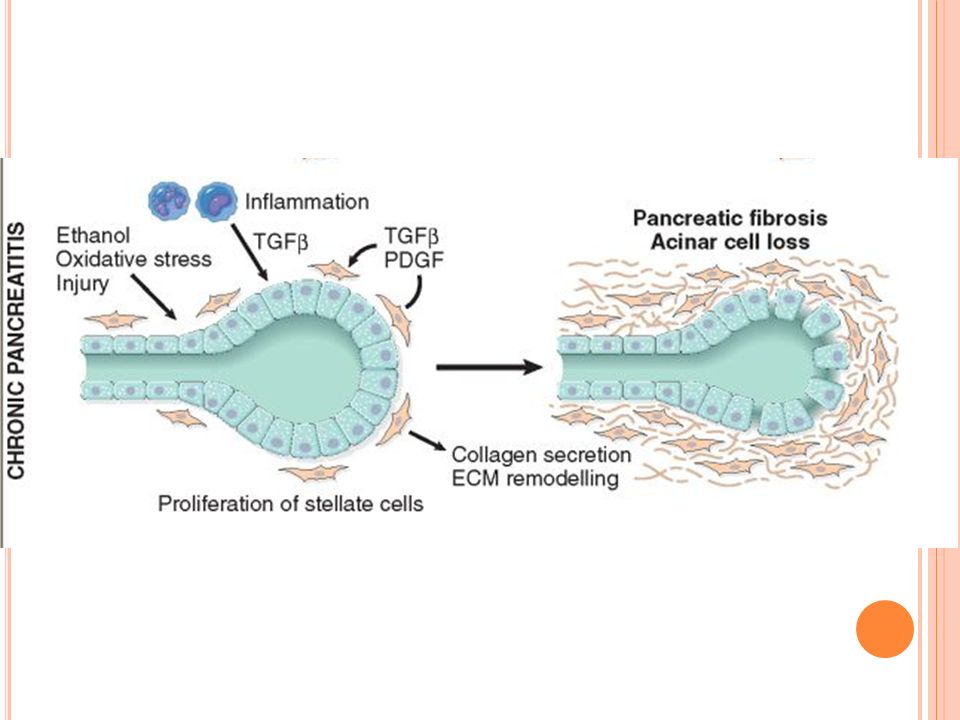
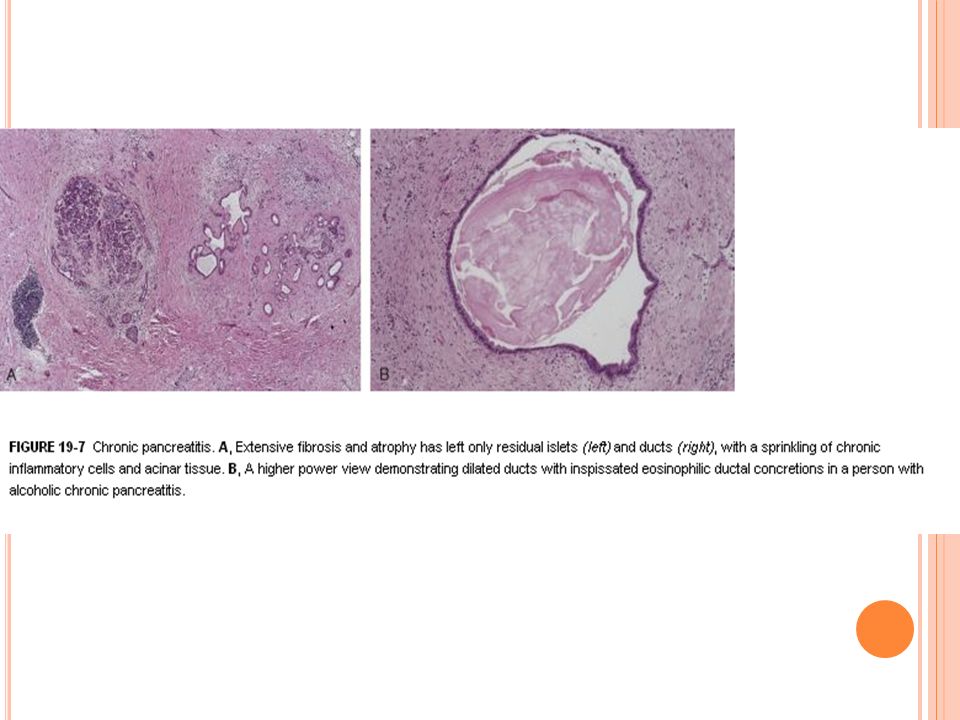
Pancreatitis crónica

73
Definición Pancreatitis crónica es una enfermedad inflamatoria del páncreas que resulta en cambios estructurales permanentes y que conducen a una insuficiencia pancreática exocrina y/o endocrina

76
Etiología Alcohol Pancreatitis hereditaria Obstrucción ductal
Enfermedades sistémicas Lupus eritematoso Fibrosis quística Hiperparatiroidismo Pancreatitis idiopáticas Mutaciones del gene de fibrosis

77
Patogénesis Depósitos proteicos ductales Isquemia Antioxidantes
Desórdenes autoinmunes

78
Síntomas Dolor abdominal Insuficiencia pancreática Exocrina Endocrina
Mala absorción de grasas Endocrina Diabetes

79
Diagnóstico diferencial
Pancreatitis aguda Cáncer de páncreas Pancreatitis auto inmune Otras

80
Complicaciones Seudoquiste Obstrucción biliar Obstrucción duodenal
Ascitis y derrame pleural Trombosis venosa Pseudoaneurisma

81
Diabetes mellitus

82
Diabetes mellitus Grupo de enfermedades crónicas con un metabolismo deficiente de carbohidratos y proteínas El defecto en la insulina se traduce en uso inadecuado de los carbohidratos e hiperglicemia 3% de la población mundial

83
Clasificación Primaria o secundaria Secundaria:
*destrucción del páncreas *endocrinopatías *drogas *gestacional Primaria: *Tipo 1(insulino dependiante): 10% *Tipo 2( no insulino dependiente): 80-90% *MODY: menos del 5%
: 10% *Tipo 2( no insulino dependiente): 80-90% *MODY: menos del 5%")
84
Diabetes mellitus Aunque los mecanismos patogenéticos sean diferentes en los diversos tipos, las complicaciones agudas y crónicas son similares Afecta 13 millones de personas en US Séptima causa de muerte en US El riesgo de desarrollas DM tipo 2 es 7% y tipo %

85
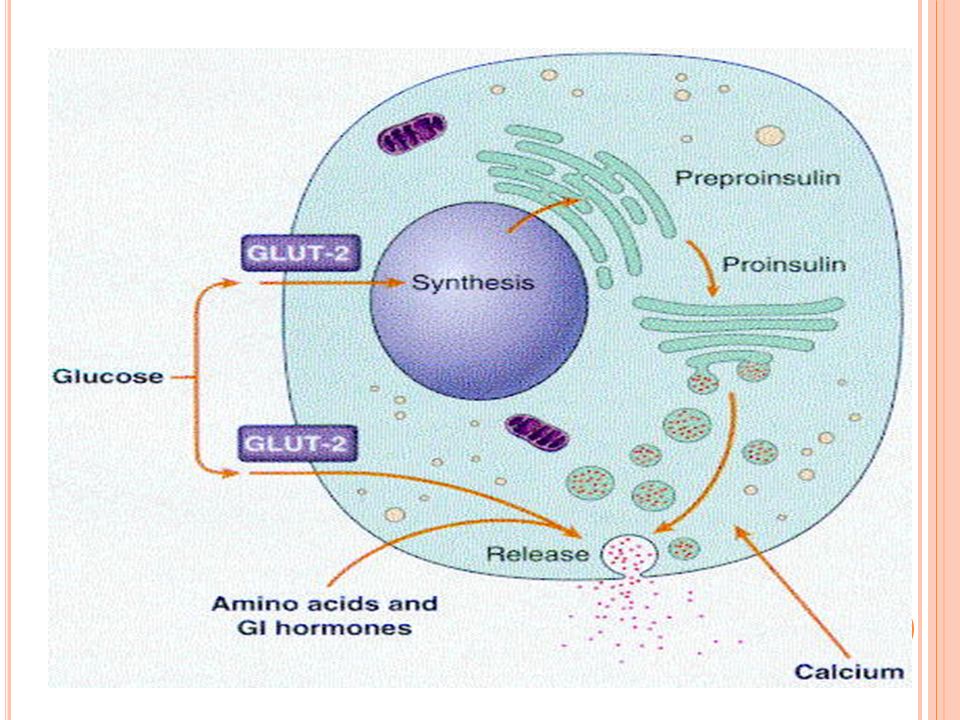
Fisiología de la insulina
La homeostasia de la glucosa tiene tres elementos: *Producción hepática *Captación de la glucosa por los tejidos *Secreción de insulina La insulina se secreta como preproinsulina y se degrada por convertasas en insulina y péptido C

87
Fisiología de la insulina
El estímulo que desencadena la liberación de insulina es la glucosa Si el estímulo persiste se sintetiza nueva Otros agentes estimulan la liberación: sulfonilureas, leu, arg y hormonas intestinales

88
Fisiología de la insulina
La insulina es necesaria para: Transporte transmembrana de glucosa Formación de glucosa en hígado y músculo Conversión de glucosa a TG’s Síntesis de ácidos nucleicos: estimula G y diferenciación en algunos tejidos Síntesis proteica *su principal función es aumentar el transporte de glucosa a ciertas células

89
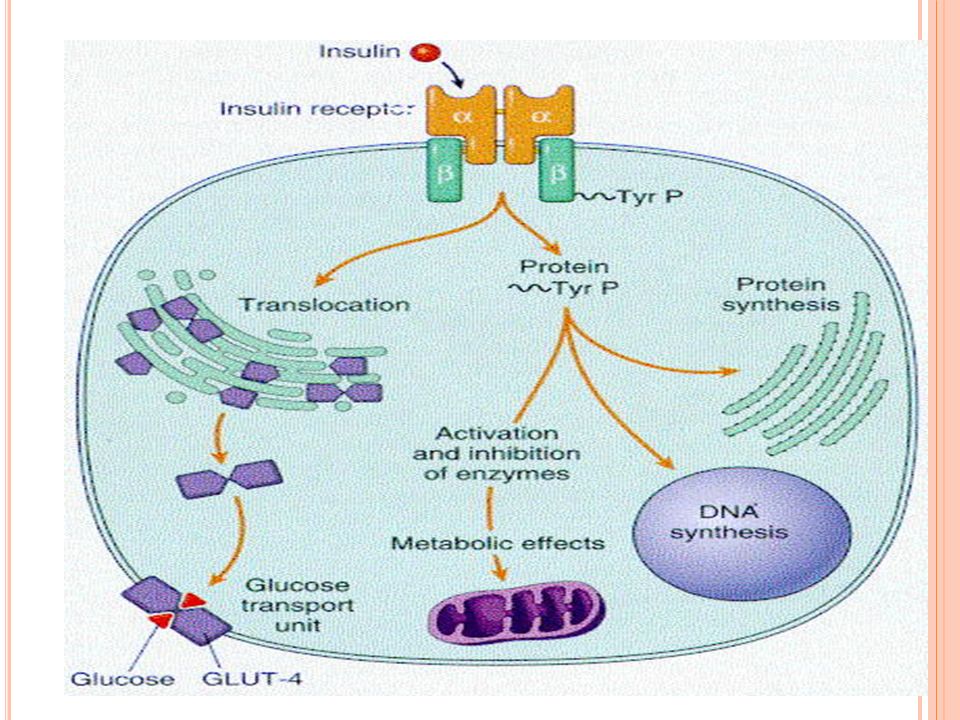
Fisiología de la insulina
Receptor de insulina: glicoproteínas con subunidades alfa y beta La activación del receptor desencadena una serie de eventos incluyendo la activación de ADN y de vías anabólicas e inhibición de vías catabólicas GLUT’s: unidades de transporte de glucosa Al activrse el receptor de insulina los GLUT’s pasan del Golgi a la membrana y facilitan la captación de la glucosa

91
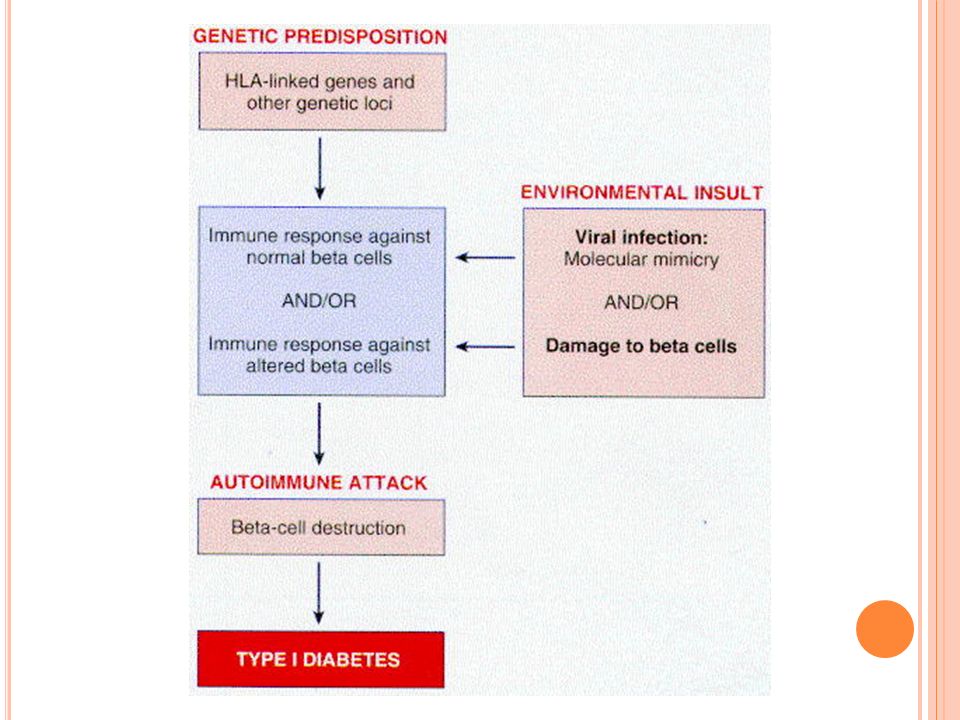
Diabetes Mellitus tipo 1: patogénesis
Ausencia severa absoluta de insulina Reducción de la masa de células B Aparece en la infancia y los pacientes requieren insulina, de otra forma desarrollan cetoacidosis y coma Hay 3 elementos para la destrucción de las células B: genético, autoinmune y ambiental

92
Diabetes Mellitus tipo 1: patogénesis
Susceptibilidad genética: más frecuente en ciertas poblaciones étnicas Al menos uno de los genes de susceptibilidad reside en el 6p21(HLA-D) 95% de los pacientes tienen HLA-DR3 ó 4 Estos genes alteran el grado de respuesta inmune en pacientes susceptibles y se genera una respuesta IL anormal Hay al menos otras 20 regiones cromosómicas involucradas
 95% de los pacientes tienen HLA-DR3 ó 4. Estos genes alteran el grado de respuesta inmune en pacientes susceptibles y se genera una respuesta IL anormal. Hay al menos otras 20 regiones cromosómicas involucradas.")
93
Diabetes Mellitus tipo 1: patogénesis
Autoinmunidad: el ataque a las células inicia muchos años antes que los sx La enfermedad se manifiesta hasta que se ha destruido 90% de la masa de células B Se observa insulitis con predominio de CD8 Autoanticuerpos: contra Ag’s intracelulares de los islotes Asociado a otros síndromes autoinmunes como Graves y tiroiditis

94
Diabetes Mellitus tipo 1: patogénesis
Ambientales: Virus: Coxsackie grupo B y otros con tropismo para las células B que en individuos susceptibles podrían desencadenar la respuesta IL anormal por antígenos virales secuestrados o similares con los de las células B Otros: niños que ingieren derivados de leche de vaca antes de los 4 meses de vida químicos

96
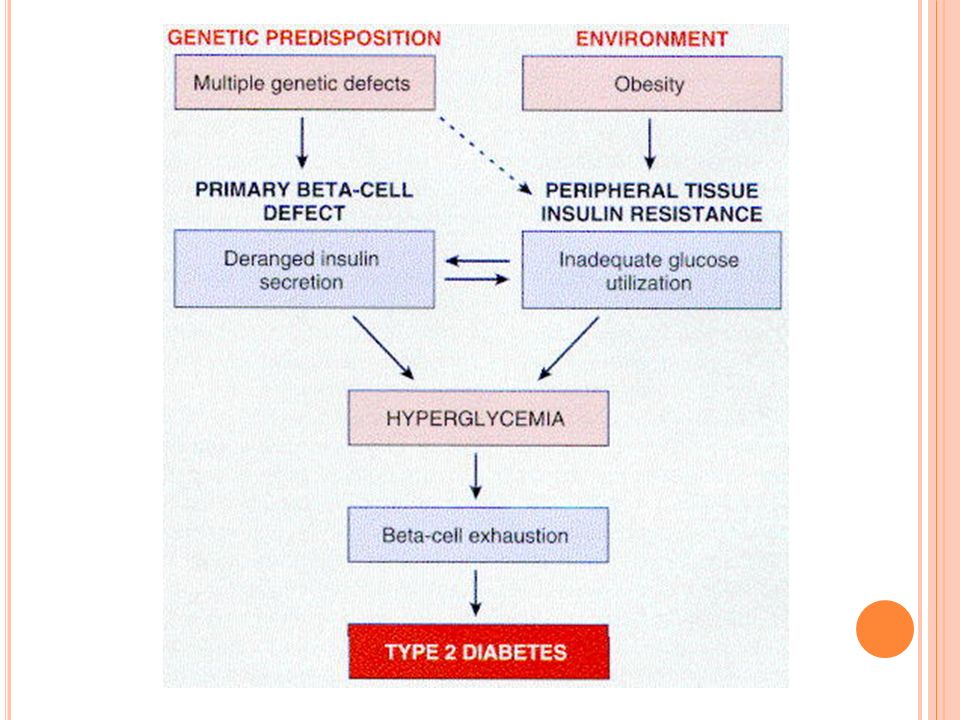
Diabetes mellitus tipo 2: patogénesis
La patogenia no está totalmente clara Los factores genéticos son aún más importantes que en la tipo 1 Los factores ambientales también afectan Es herencia multifactorial El elemento clave es la disminución a la respuesta periférica a la insulina, el deterioro en la secreción también existe

97
Diabetes mellitus tipo 2 patogénesis
Deterioro en la secreción: al principio los niveles de insulina no bajan pero los pulsos normales de secreción se pierden Posteriormente aparece leve a moderado déficit de insulina Esto parece ser causa de la hiperglicemia crónica que agota la fn de la células B

98
Diabetes mellitus tipo 2 patogénesis
Resistencia a la insulina: respuesta reducida de los tejidos periféricos a insulina Esta condición no es exclusiva de la DM: presente en embarazo y obesidad aún en ausencia de DM Hay deterioro en las señales postreceptor Hay reducción de la síntesis y translocación de las GLUT’s

99
Diabetes mellitus tipo 2: patogenia
El déficit en la respuesta a la insulina lleva a que no se dispone adecuadamente de la glucosa, con hiperglicemia crónica y estímulo persistente de las células B Obesidad: 80% de los IDDM son obesos, principalmente intraabdominalla grasa abdominal libera AG al hígado y no responde a los efecos moduladores de la insulina La pérdida de peso revierte el defecto Amylina: proteína que se secreta con la insulina y que se acumula como amiloide en DM

101
MODY Tienen un grupo heterogéneo de defectos genéticos en la función de las células B AD Inicia antes de los 25 años Peso normal, sin auto ac’s, sin resistencia a insulina Se dividen en 4 grupos según el defecto

102
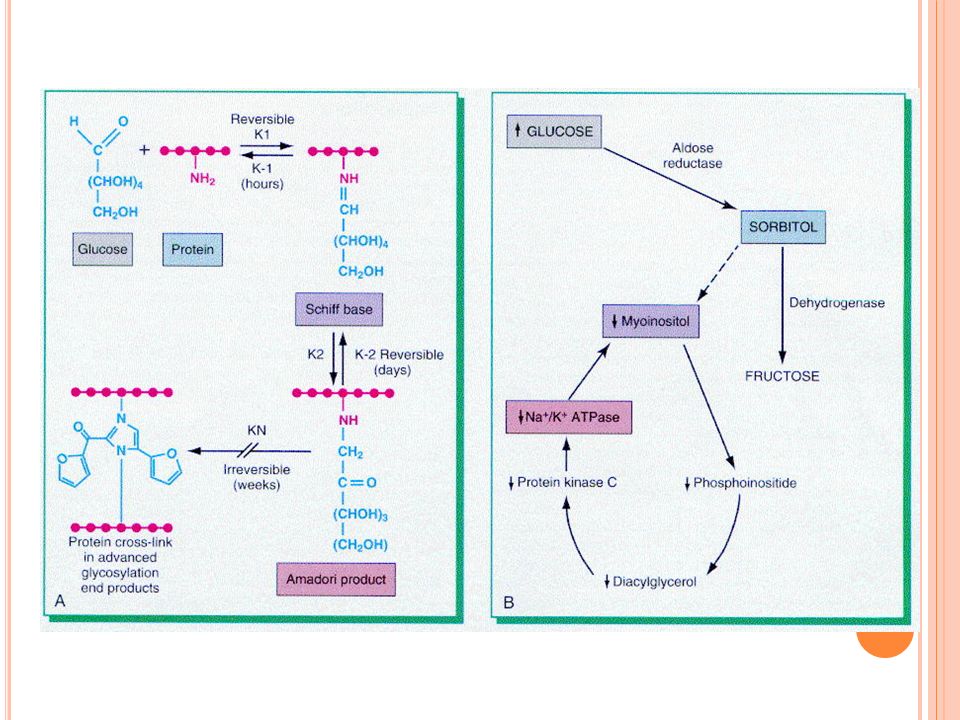
Patogénesis de las complicaciones de la DM
Son consecuencia principalmente de la hiperglicemia Glicosilación no enzimática: la glucosa se une a los grupos amino de las proteínas, el grado de glicosilación es proporcional a la glicemia Hb glicosilada para valorar control de glicemia La glicosilación no revierte sino que sufre cambios crónicos que llevan a la formación de productos terminales de glicosilación(PTG) que se acumulan en las paredes vasculares
 que se acumulan en las paredes vasculares.")
103
Patogénesis de las complicaciones de la DM
PTG:se depositan en proteínas como la colágena, esta atrapa proteínas intersticiales no glicosiladas y LDL, acelerando la aterogénesis PTG:se unen a receptores en diversas células lo que induce cambios como migración de monocitos, liberación de citocinas, aumento de síntesis de matriz extracelular, etc. Hiperglicemia intracelular: en tejidos que no requieren insulina para captar glucosa

105
Patogénesis de las complicaciones de la DM
En estos tejidos como SN se metaboliza el exceso de glucosa a sorbitol y fructosa. Esto aumenta P osm intracelular y da edema intracelular Esto da opacidad y edema del cristalino y deteriora la función de las bombas iónicas llevando a daño en células de Schwann y pericitos produciendo neuropatía y microaneurismas en la retina

107
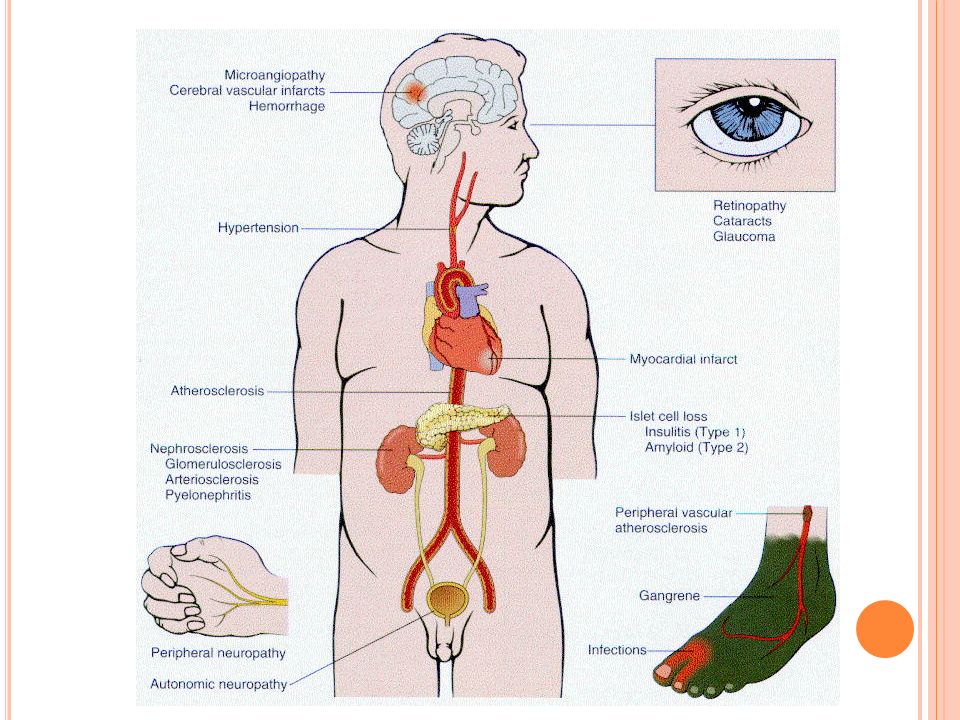
Morfología de la Diabetes Mellitus
Páncreas: principalment en la tipo 1 *disminución en número y tamaño de los islotes de Langerhans *Insulitis *En la tipo 2 hay leve reducción del número de islotes y depósitos de amiloide y fibrosis *En RN de madres diabéticas se encuentra aumento del número de islotes en respuesta a la hiperglicemia materna: nesiodioblastosis fetal

108
Morfología de la Diabetes Mellitus
Sistema vascular: *ateroesclerosis acelerada *el infarto de miocardio es la causa de muerte más común en diabéticos *gangrena de extremidades inferiores:100 veces más frecuente en DM que en el resto de la población +los pacientes tienen dislipidemia con frecuencia, glicosilación y atrapamiento de lipoproteínas, aumento de adhesividad plaquetaria y incidencia alta de HTA

109
Morfología de la Diabetes Mellitus
Arterioloesclerosis hialina: asociada a HTA y envejecimiento. Es frecuente en DM pero no específica Microangiopatía: engrosamiento difuso de membranas basales capilares en piel, músculo, retina, glomérulos y médula renal y cualquier otro órgano Los vasos a pesar de ser más gruesos son más permeables a las proteínas

111
Morfología de la Diabetes Mellitus
Nefropatía diabética:segunda causa de muerte en diabéticos Glomérulos: engrosamiento de m.basal, gloméruloesclerosis focal y difusa con proliferación mesangial Nodular: Kimmelstiel-Wilson Vasos: ateroesclerosis y arterioloesclerosis Pielonefritis aguda y crónica Papilitis necrotizante:

112
Morfología de la Diabetes Mellitus
Oculares: retinopatía, cataratas, glaucoma En conjunto constutiyen la cuarta causa de ceguera en US Neuropatía: neuropatía simétrica periférica de miembros inferiores que afecta tanto funciones motoras como sensitivas pero más particularmente sensitiva

113
Características clínicas
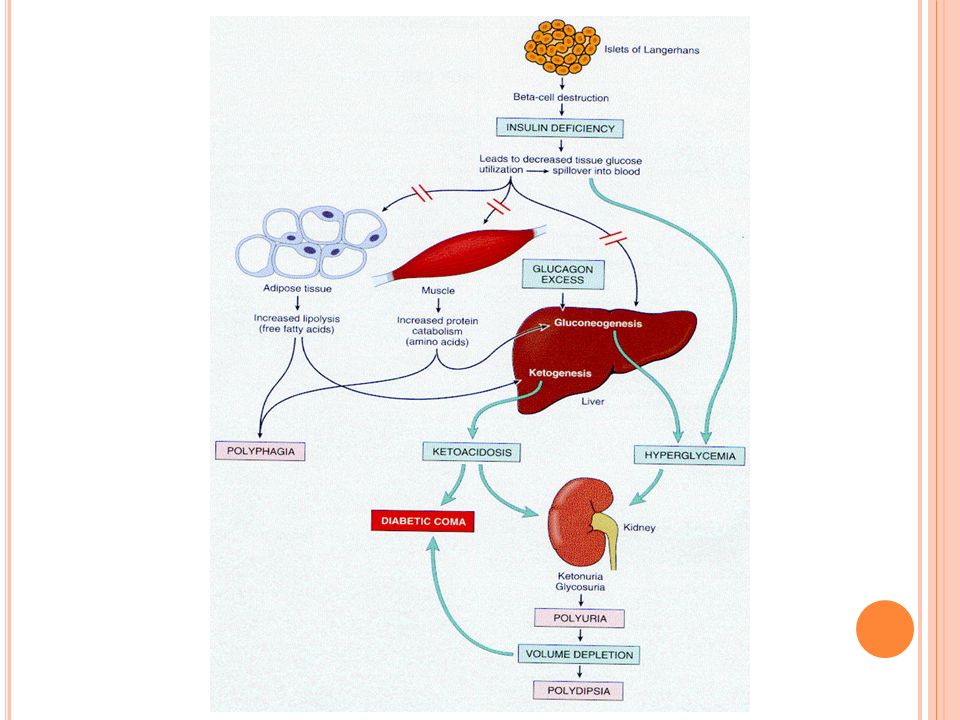
Tipo 1: se inocia en pacientes jóvenes con polidipsia, poliuria, polifagia y cetoacidosis Se produce un estado catabólico de proteínas y grasa. Los aa’s de la proteolisis se usan para sintetizar glucosa Se produce pérdida de peso y debilidad La cetoacidosis se produce por severa depleción de insulina y exceso de glucagon. Esto lleva a degradación de grasa y exceso de ácidos grasos que se oxidan en el hígado a cuerpos cetónicos

114
Características clínicas
El glucagon acelera la oxidación de los ácidos grasos Se produce cetonemia y cetonuria Si hay deshidrataciónd se deteriora la excreción de H+ y se produce cetoacidosis Son pacientes más susceptibles a las infeccones y estas aumentan los requerimientos de glucosa y desencadenan descompensaciones

116
Características clínicas
Los DM tipo 2 inician a edad más tardía y son obesos No hacen cetosis, las descompensaciones son hiperosmolares Las infecciones causan la muerte al 5% de los pacientes y más graves Una descompensación puede seguir a un proceso trivial como una infección en un pie Los pacientes con DM tipo 1 tienen más riesgo de morir de la enfermedad

117
Gracias

Presentaciones similares