Descargar la presentación
La descarga está en progreso. Por favor, espere

1
PATOLOGÍA DE LA HERENCIA
Dr. Minor Vargas Baldares Médico Patólogo Hospital San Juan de Dios

2
www.ncbi.nim.nihgov: National Library of Medicine. Referencia general
Índice de genes, rasgos y enfermedades genéticas Trastornos genéticos: Clínica y laboratorio American Society of Human Genetics Aspectos sanitarios, clínicos, legales, sociales y éticos

3
TRASTORNOS GENÉTICOS Los primeros trastornos genéticos que se identificaron fueron los “errores congénitos del metabolismo” (Sir Archibald Garrod, 1908), tratados parcialmente con dieta y consejería genética
, tratados parcialmente con dieta y consejería genética.")
4
Frecuencia: 67% de todas las personas (a lo largo de su vida)
“Clásicas” (monogénicas) Herencia multifactorial (poligénicas) Enfermedades cardiovasculares Cáncer: acumulación de mutaciones en células somáticas Son mucho más frecuentes y se presentan sobre todo en adultos
 Herencia multifactorial (poligénicas) Enfermedades cardiovasculares. Cáncer: acumulación de mutaciones en células somáticas. Son mucho más frecuentes y se presentan sobre todo en adultos.")
5
Abortos espontáneos: 10% de embarazos
50% anormalidades cromosómicas ?% errores genéticos detectables La mayoría de las enfermedades cromosómicas son incompatibles con la vida. Sólo unas pocas trisomías (13, 18, 21, Y, X) y micro delecciones sobreviven con fenotipos específicos e inespecíficos
 y micro delecciones sobreviven con fenotipos específicos e inespecíficos.")
6
Recién nacidos 1% con anormalidades cromosómicas
Ingresos hospitalarios en Pediatría 25% determinados por un trastorno monogénico o multifactorial Más de 50% de las enfermedades crónicas infantiles son genéticas o influenciadas por una predisposición genética 34% de las muertes infantiles hospitalarias se asocian con trastornos genéticos

7
Las enfermedades monogénicas son raras pero son muchas y variadas por lo que juntas constituyen un grupo significativo Menores de 25 años 5% tienen enfermedad genética seria

8
PREVENCIÓN Parejas de riesgo: Análisis genéticos para detección de mutaciones específicas Sensibilidad a medicamentos. Cada persona responde de manera distinta a cada fármaco. En el futuro se podrá adaptar el tratamiento a las variaciones individuales del genoma de cada paciente

9
DIAGNÓSTICO El diagnóstico prenatal se puede efectuar con marcadores plasmáticos maternos, ecografía fetal, amniocentesis (10-18 semanas) y biopsia de vellosidades coriales (10-12 semanas) El diagnóstico preimplantatorio (DGP) se efectúa en blastómeros de embriones iniciales para seleccionar e implantar únicamente los embriones no afectados Actualmente la selección neonatal con espectrometría masiva permite la detección de metabolitos anormales con un solo análisis muy económico
 y biopsia de vellosidades coriales (10-12 semanas) El diagnóstico preimplantatorio (DGP) se efectúa en blastómeros de embriones iniciales para seleccionar e implantar únicamente los embriones no afectados. Actualmente la selección neonatal con espectrometría masiva permite la detección de metabolitos anormales con un solo análisis muy económico.")
10
TRATAMIENTO Ya se ha iniciado el tratamiento con enzimas modificadas lo cual exige un diagnóstico oportuno La reposición génica y el uso de células madre no han dado resultados positivos a la fecha

11
PROFESIONALES EN GENÉTICA
Especialistas en Genética: Médicos con especialidad (residencia hospitalaria en Genética) Asesor en Genética: Biológicos, enfermeras, con maestría universitaria Genetista de Laboratorio: Médico con mínimo 2 años de trabajo en investigación en genética
 Asesor en Genética: Biológicos, enfermeras, con maestría universitaria. Genetista de Laboratorio: Médico con mínimo 2 años de trabajo en investigación en genética.")
12
Atención del Paciente Médico general Pediatra o internista
Médico especialista Médico genetista

13
ÉTICA EN GENÉTICA “Nada es tan personal como el material genético de cada persona” La discriminación genética debería ser ilegal: un diagnóstico genético personal o familiar no debería afectar la contratación laboral, suscribir un seguro de gastos médicos o de vida, o un pronunciamiento judicial

14
La información genética requiere del mayor grado de confidencialidad que impida cualquier estigma para el paciente o su familia El interés de cualquier análisis genético siempre debe centrarse en el interés del niño paciente y no de sus padres o la colectividad Las pruebas de diagnóstico molecular sólo están indicadas en casos de síndromes malformativos, retraso mental y otras discapacidades en las que exista claro el beneficio para el paciente

15
Recomendaciones Éticas de la American Academy of Pediatrics (AAP) 2001
Las pruebas establecidas de detección selectiva deberían revisarse y evaluarse periódicamente para permitir que se modifique el programa. Su introducción a nivel neonatal debería realizarse mediante protocolos de investigación cuidadosamente controlados Análisis genéticos y esfuerzos diagnósticos o terapéuticos para los niños, requieren un proceso de consentimiento informado de los progenitores y la conformidad de los niños mayores. La frecuencia de los rechazos informados debería controlarse. Es obligatorio investigar para mejorar la efectividad del consentimiento informado para la detección selectiva neonatal

16
La AAP no respalda el uso generalizado de los análisis o la detección selectiva de los portadores en niños o adolescentes. Los riesgos y beneficios de la detección selectiva de portadores en la población pediátrica deberían evaluarse en los ensayos clínicos controlados antes de ofertarla a gran escala. La detección de portadores en adolescentes embarazadas o en adolescentes que deseen quedar embarazadas puede ser apropiada Los análisis genéticos de trastornos que se inicien en la edad adulta deberían diferirse hasta dicha época o hasta que los adolescentes interesados en el análisis hayan desarrollado las capacidades maduras de toma de decisiones. Los análisis genéticos de los niños y adolescentes para predecir trastornos de inicio tardío son inadecuados si no se ha demostrado que la información genética reduzca la morbilidad y mortalidad

17
Es posible que la detección selectiva y los análisis genéticos no sean bien comprendidos, los pediatras deben proporcionar a los progenitores la información y asesoramiento necesarios sobre el conocimiento genético y posibilidades terapéuticas, posibles perjuicios que pueden causarse al adquirir cierta información genética (daño psicológico, estigmatización y discriminación), así como sobre los trastornos médicos y la discapacidad. Los pediatras pueden recibir ayuda gracias a la colaboración de genetistas, asesores genéticos y profesionales sanitarios de medicina neonatal La AAP respalda la expansión de ofertas educativas sobre genética humana para estudiantes de medicina, residentes y médicos en ejercicio, así como la expansión de programas de formación para profesionales de genética

18
GENOMA HUMANO Y GENÓMICA
La genómica estudia todos los genes del genoma y sus interacciones, el arreglo estructural del ADN en el análisis de tumores y sobre todo la posibilidad de descifrar la compleja relación entre el genoma y el ambiente en las enfermedades genéticas multifactoriales

19
Hallazgos Sorprendentes de la Genómica
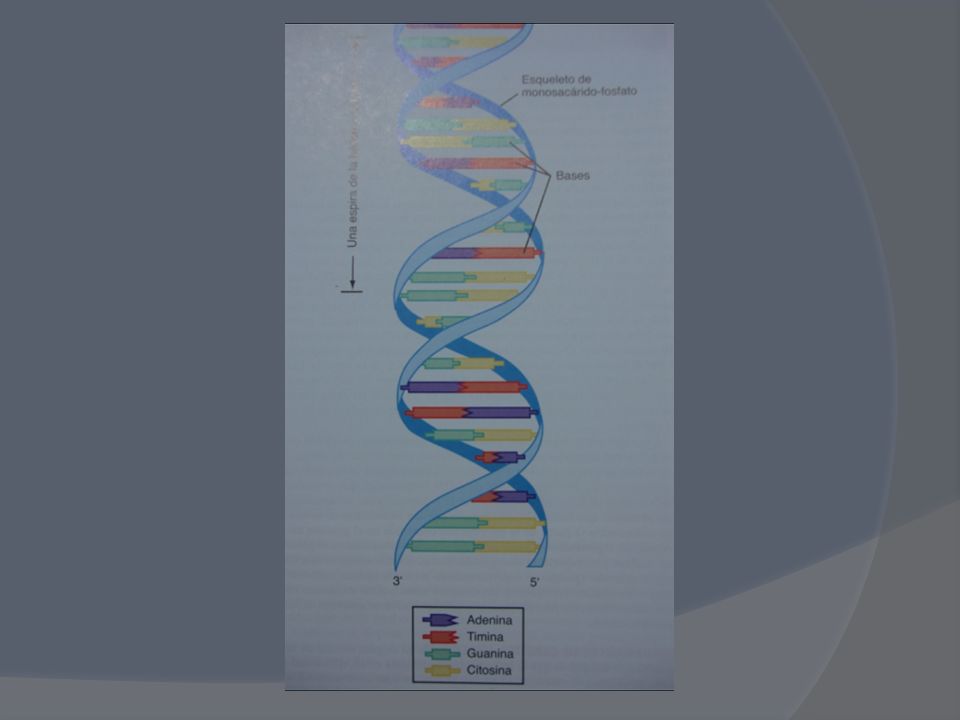
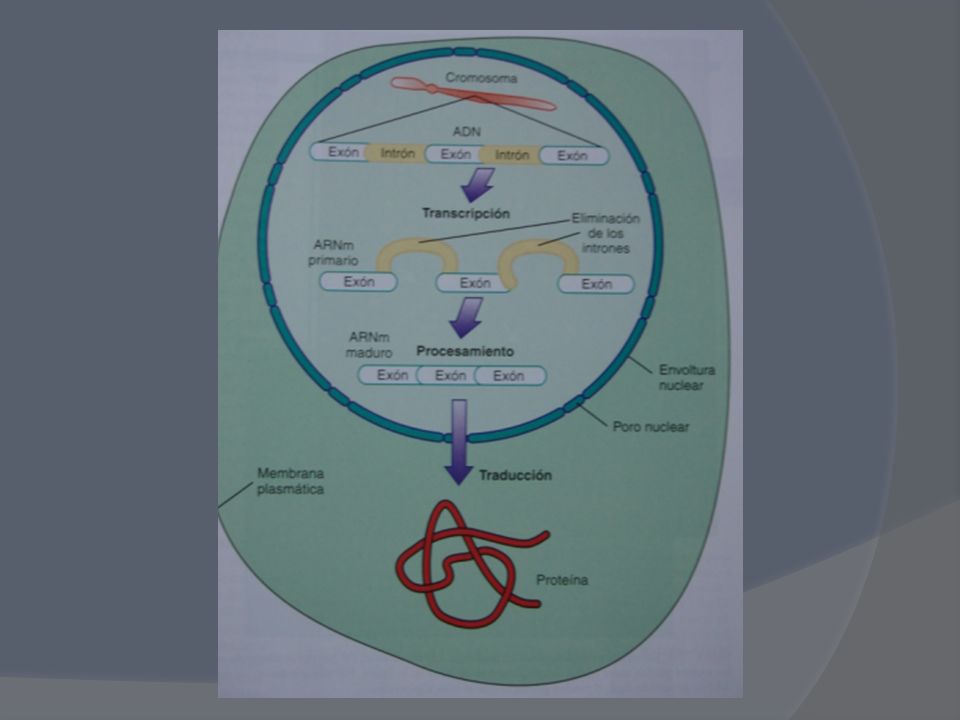
Menos del 2% de todo el genoma codifica proteínas Más del 50% del genoma lo constituyen bloques de nucleótidos repetidos cuya función permanece en el misterio El ser humano posee solamente genes o unidades individuales de herencia de todos los rasgos genéticos, organizados en los largos segmentos del ADN dispersos en el núcleo (eucromatina) y que durante la mitosis se concentran junto con proteínas para formar los cromosomas.
 y que durante la mitosis se concentran junto con proteínas para formar los cromosomas.")
20
Además de los 46 cromosomas del núcleo existe un “cromosoma” o ADN en las mitocondrias que contienen pares de bases completamente secuenciada a partir de la cual se forman las mitocondrias. El ADN mitocondrial y por lo tanto la mitocondria son de origen materno, puesto que los espermatozoides no pueden aportar mitocondrias al óvulo fertilizado

21
Los seres humanos compartimos el 99,9% de las secuencias del ADN
Los seres humanos compartimos el 99,9% de las secuencias del ADN. La gran diversidad (ningún ser humano es igual a otro) se halla codificada en sólo un 0,1% de nuestro ADN: aquí reside también nuestra predisposición a enfermedades y la respuesta y resistencia a los agentes tóxicos y químicos, biológicos y medicamentosos e incluso psicológicos. Sin embargo ese 0,1% constituye alrededor de de pares de bases, casi 200 veces más que las del ADN mitocondrial
 se halla codificada en sólo un 0,1% de nuestro ADN: aquí reside también nuestra predisposición a enfermedades y la respuesta y resistencia a los agentes tóxicos y químicos, biológicos y medicamentosos e incluso psicológicos. Sin embargo ese 0,1% constituye alrededor de de pares de bases, casi 200 veces más que las del ADN mitocondrial.")
24
TRASTORNOS GENÉTICOS

25
TRASTORNOS DE HERENCIA MENDALIANA
El número de trastornos mendelianos ha aumentado en grandes proporciones Se estima que todo individuo es portador de 5 a 8 genes anormales La mayoría de estos trastornos son recesivos 80-85% son familiares, el resto son mutaciones “de novo” Expresión parcial en el heterozigota y total en el homozigota

26
Codominancia: Expresión total en heterozigotas
Pleiotropismo: Una simple mutación produce muchos efectos Heterogeneidad: Un fenotipo puede ser producido por numerosas mutaciones diferentes Polimorfismo simple: Cambio en un nucleótido sin consecuencias, ocurre en más del 1% de la población

28
TRASTORNOS AUTOSÓMICOS DOMINANTES
Determinado por la presencia de un gen anómalo en uno de los autosomas Se manifiestan en el estado heterozigota Por lo menos un progenitor está afectado Trasmisión es vertical en todas las generaciones Riesgo de recurrencia 50% Hombres y mujeres se afectan por igual Los individuos no afectados no transmiten la enfermedad

29
La trasmisión de varón a varón afirma la herencia autosómica dominante
Si la enfermedad reduce la fertilidad, aumenta la proporción de mutaciones “de novo” El fenotipo no sólo se refiere a manifestaciones físicas sino a las conductuales y de laboratorio Penetrancia reducida Expresividad variable Mutaciones somáticas El inicio de la enfermedad es retardado, usualmente en el adulto

30
TRASTORNOS AUTOSÓMICOS RECESIVOS
Se manifiestan en el estado homozigota Implica la existencia de mutaciones en ambas copias del gen Trasmisión horizontal: suele haber generaciones sin casos Riesgo de recurrencia 25% Los padres no suelen estar afectados

31
Varones y hembras se afectan por igual (algunos rasgos tienen expresión diferente)
Mayor frecuencia de consanguinidad La expresión del defecto suele ser más uniforme La penetrancia completa es más común La enfermedad suele iniciarse a edades tempranas Incluyen la mayoría de los errores congénitos del metabolismo Herencia pseudodominante

32
TRASTORNOS LIGADOS AL SEXO
Varones se ven afectados con más frecuencia Las mujeres portadoras no suelen verse afectadas Todos son ligados al cromosoma X Casi todos son recesivos Varones con mutaciones en el cromosoma Y son infértiles Riesgo de recurrencia: 25% Los trastornos se expresan en el varón Los varones no trasmiten la enfermedad a sus hijos, pero todas sus hijas son portadoras Hijas de madres heterozigota tienen 50% de probabilidad de recibir el gen mutante Es posible que una mujer exprese el fenotipo si el alelo normal es inactivado en la mayoría de las células

33
ENFERMEDADES AUTOSÓMICAS DOMINANTES
SISTEMA ENFERMEDAD Nervioso Enfermedad de Huntigton Neurofibromatosis Distrofia miotónica Esclerosis tuberosa Urinario Enfermedad renal poliquística Gastrointestinal Poliposis colónica familiar Hematopoyético Esferocitosis hereditaria Enfermedad de von Willebrand Esquelético Síndrome de Marfan Síndrome de Ehlers-Danlos (algunas variantes) Osteogénesis imperfecta Acondroplasia Metabólico Hipercolesterolemia familiar Porfiria intermitente aguda
 Osteogénesis imperfecta. Acondroplasia. Metabólico. Hipercolesterolemia familiar. Porfiria intermitente aguda.")
34
TRASTORNOS AUTOSÓMICOS RECESIVOS
SISTEMA ENFERMEDAD Metabólico Fibrosis quística Fenilcetonuria Galactosemia Homocistinuria Enfermedades por almacenamiento lisosomales Deficiencia de α-antitripsina Enfermedad de Wilson Hemocromatosis Enfermedades por almacenamiento de glucógeno Hematopoyético Anemia de células falciformes Talasemias Endocrino Hierplasia suprarrenal congénita Esquelético Síndrome de Ehlers-Danlos (algunas variantes) Alcaptonuria Nervioso Atrofias musculares neurogénicas Ataxia de Friedreich Atrofia muscular espinal
 Alcaptonuria. Nervioso. Atrofias musculares neurogénicas. Ataxia de Friedreich. Atrofia muscular espinal.")
35
ENFERMEDADES RECESIVAS LIGADAS AL CROMOSOMA X
SISTEMA ENFERMEDAD Músculoesquelético Distrofia muscular de Duchene Sangre Hemofilia A y B Enfermedad granulomatosa crónica Deficiencia de glucosa-6-fosfato deshidrogenasa Inmunitario Agammaglobulinemia Síndrome de Wiskott-Aldrich Metabólico Diabetes insípida Síndrome de Lesh-Nyhan Nervioso Síndrome del cromosoma X frágil

36
BASES BIOQUÍMICAS Y MOLECULARES DE ALGUNAS ENFERMEDADES MENDELIANAS
TIPO/FUNCIÓN DE LA PROTEÍNA EJEMPLO ENFERMEDAD Enzima Fenilalanina hidroxilasa Hexosaminidasa Adenosina desaminasa Fenilcetonuria Enfermedad de Tay-Sachs Inmunodeficiencia combinada grave Inhibidor enzimático α-antitripsina Enfisema y enfermedad hepática Receptor Receptor de lipoproteína de baja densidad Receptor de vitamina D Hipercolesterolemia familiar Raquitismo resistente a la vitamina D

37
TIPO/FUNCIÓN DE LA PROTEÍNA
EJEMPLO ENFERMEDAD Transporte Oxígeno Iones Hemoglobina Regulador de la conductancia transmembrana de la fibrosis quística Talasemia, anemia de células falciformes Fibrosis quística Estructural Extracelular Membrana celular Colágeno Fibrina Distrofina Espectrina, anquirina o proteína 4.1 Osteogénesis imperfecta, síndrome de Ehlers-Danlos Síndrome de Marfan Distrofia muscular de Duchene/Becher Esferocitosis hereditaria Hemostasia Factor VIII Hemofilia A Regulación del crecimiento Proteína Rb Neurofibromina Retinoblastoma hereditario Neurofibromatosis tipo 1

38
HERENCIA MULTIFACTORIAL
El riesgo depende del número de mutantes heredades (parientes con expresiones severas) El riesgo de recurrencia (2-7%) el mismo para todos los parientes en primer grado El riesgo para gemelos idénticos es 20-40% En embarazos subsecuentes el riesgo aumenta si el anterior estuvo afectado (7-9%) El riesgo de expresión del rasgo discontinuo (diabetes) ocurrirá solamente cuando la influencia sumatoria de genes y ambiente superen un umbral determinado
 El riesgo de recurrencia (2-7%) el mismo para todos los parientes en primer grado. El riesgo para gemelos idénticos es 20-40% En embarazos subsecuentes el riesgo aumenta si el anterior estuvo afectado (7-9%) El riesgo de expresión del rasgo discontinuo (diabetes) ocurrirá solamente cuando la influencia sumatoria de genes y ambiente superen un umbral determinado.")
39
SÍNDROME DE MARFAN: Fibrilinopatía Tipo 1
70-85% son familiares Autosómico dominante con expresión variable Defecto en fibrilina 1 (principal componente de microfibrillas en que se deposita la elastina) Más de 500 mutaciones del gene FBN1 Extremidades largas. Hiperextensión. Dolicocefalia. Pliegues supraorbitarios. Pectus excavatum (Abraham Lincoln). Subluxación y dislocación de cristalino (ectopia lentis). Prolapso de válvula mitral. Dilatación de aorta ascendente
 Más de 500 mutaciones del gene FBN1. Extremidades largas. Hiperextensión. Dolicocefalia. Pliegues supraorbitarios. Pectus excavatum (Abraham Lincoln). Subluxación y dislocación de cristalino (ectopia lentis). Prolapso de válvula mitral. Dilatación de aorta ascendente.")
40
EHLERS – DANLOS Grupo heterogéneo con el mismo defecto en la síntesis de colágena 3 patrones mendelianos (dominante-recesivo-ligado al sexo) 6 variantes clínicas (clásica, hipermovilidad, vascular, cifoescoliosis, artrocalasia, dermatosparaxis) El gene es muy largo con más de 900 mutaciones identificadas en el cromosoma 19
 El gene es muy largo con más de 900 mutaciones identificadas en el cromosoma 19.")
41
HIPERCOLESTEROLEMIA FAMILIAR
¨Enfermedad de receptor¨ para LDL, necesario para el transporte y metabolismo del colesterol (pérdida del control de retroalimentación y aterosclerosis prematura) Es el trastorno mendeliano más frecuente (heterozigoto con una mutante, 1:500) Heterozigotos: 2-3 veces más colesterol desde el nacimiento Homozigotos: 5-6 veces más colesterol desde el nacimiento
 Es el trastorno mendeliano más frecuente (heterozigoto con una mutante, 1:500) Heterozigotos: 2-3 veces más colesterol desde el nacimiento. Homozigotos: 5-6 veces más colesterol desde el nacimiento.")
42
TAY – SACHS Deficiencia de hexosaminidasa A
Acumulación de gangliósidos en neuronas Nacimiento normal. Síntomas a los 6 meses (incoordinación, retardo mental, flacidez, ceguera, demencia). Muerte en 2-3 años
. Muerte en 2-3 años.")
43
NIEMANN – PICK (A, B y C) Deficiencia de esfingomielinasa
Tipo A: Infantil, severo, neurológico y visceral: muerte en 3 años Tipo B: Organomegalia sin afección encefálica, muerte en el adulto Tipo C: Hidrops fetalis, aborto, ataxia, parálisis supranuclear

44
GAUCHER Deficiencia de glucocerebrosidasa Tesaurismosis más frecuente
Depósitos de cerebrósidos en SRE Tipo I: (99%) Crónico no neuropático. Esplenomegalia. Alcanzan la edad senil Tipo II: Agudo neuropático, infantil Tipo III: Intermedio entre I y II
 Crónico no neuropático. Esplenomegalia. Alcanzan la edad senil. Tipo II: Agudo neuropático, infantil. Tipo III: Intermedio entre I y II.")
45
GLUCOGENOSIS 10 tipos Tipo I (von Gierke): Deficiencia de glucosa-6 fosfatasa. Depósitos en el hígado Tipo II (Pompe): Deficiencia de glucosidasa lisosomal. Depósitos generalizados y en corazón Tipo V (McArdle): Deficiencia de fosforilasa. Depósitos en músculo esquelético
: Deficiencia de glucosidasa lisosomal. Depósitos generalizados y en corazón. Tipo V (McArdle): Deficiencia de fosforilasa. Depósitos en músculo esquelético.")
46
NEUROFIBROMATOSIS

47
SÍNDROME DE DOWN Trastorno cromosómico más común 1:700 (95% con trisomía 21, 4% con traslocación Robertsoniana y 1% mosaicos) La causa más común de retardo mental Edad materna: <20 1:1550, >45 1:25 Causa ovular: 95% IQ 25-50: 80% (gentiles, tímidos, dóciles) Malformaciones congénitas cardiacas: 40% Leucemia aguda: más frecuente Alzheimer: Todos después de 40 años Respuestas inmunes anormales Edad promedio: 47 años (25 en 1983)
 Malformaciones congénitas cardiacas: 40% Leucemia aguda: más frecuente. Alzheimer: Todos después de 40 años. Respuestas inmunes anormales. Edad promedio: 47 años (25 en 1983)")
48
SÍNDROME DE KLINEFELTER
47XXY (82%), mosaico (15%) 1:500 en nacimientos masculinos Hábitus eunucoide. Hipogonadismo. Ginecomastia. Infertilidad o azoospermia IQ disminuido sin retardo mental Elevación de HFS y estradiol. Disminución de testosterona Ca de mama (20 veces más frecuente) LES
, mosaico (15%) 1:500 en nacimientos masculinos. Hábitus eunucoide. Hipogonadismo. Ginecomastia. Infertilidad o azoospermia. IQ disminuido sin retardo mental. Elevación de HFS y estradiol. Disminución de testosterona. Ca de mama (20 veces más frecuente) LES.")
49
SÍNDROME DE TURNER 45X0 (57%), 45XX (14%), mosaicos (29%)
1:2000 nacimientos femeninos Edema de manos y pies. Cuello palmeado (higroma coli) Estatura baja, tórax ancho, hipogonadismo, amenorrea Enfermedades congénitas cardiacas Estado mental normal Hipotiroidismo autoinmune (50%). Intolerancia a la glucosa. Obesidad Menopausia antes de la menarca
 Estatura baja, tórax ancho, hipogonadismo, amenorrea. Enfermedades congénitas cardiacas. Estado mental normal. Hipotiroidismo autoinmune (50%). Intolerancia a la glucosa. Obesidad. Menopausia antes de la menarca.")
Presentaciones similares

>")


















