Descargar la presentación
La descarga está en progreso. Por favor, espere

1
Patología General: Alteraciones Genéticas
Dr. Ignacio F. Gutiérrez Mejía Medico Patólogo UPSJB -2013

2
Genoma Humano : ADN : 23 pares de cromosomas en el núcleo.
22 cromosomas autosómicos y un par determinante del sexo XX, XY

3
1953- ADN 2000- ADN 1869- ADN

5
MUTACION Cambio permanente del ADN
Células Germinales, provoca las Enfermedades Hereditarias Células somáticas, se relaciona a Cáncer y Malformaciones congénitas. O3 categorías de mutaciones : Genoma, Cromosoma y génicas submicroscopicas

6
MUTACION MUTACION GENOMA TRISOMIA MONOSOMIAS CROMOSOMA
La mayoría incompatible con la vida Cambios estructurales génicas submicroscopicas La mayoría asociada a enfermedades hereditarias

7
Mutación por desplazamiento
Mutación Puntual Sustitución de base de nucleótido Delecion uno o dos pares de bases son insertados o delecionados del ADN Alteración en la lectura de la Cadena de ADN Mutación por desplazamiento Deleciones y alteraciones en la lectura de la cadena de ADN

8
Desordenes Citogeneticos

9
Desordenes Citogeneticos
Enfermedad Genéticas Causada por alteraciones, mutaciones del ADN Si se producen en los gametos son heredables Enfermedad Congénita Está presente desde el nacimiento

10
ENFERMEDADES MENDELIANAS.
Resultado de mutaciones expresadas en genes únicos de gran efecto. Un individuo es portador entre 5 y 8 genes deletéreos, la mayoría son recesivos y no tienen efecto fenotípicos graves % son mutaciones familiares. El resto corresponde a nuevas mutaciones adquiridas. Algunas mutaciones producen expresión parcial en el heterocigoto, y completa en el homocigoto 10

11
ENFERMEDADES MENDELIANAS
En la anemia falciforme se produce substitución de la HbA por la HbS en el heterocigoto, pero en el homocigoto toda la hemoglobina en S. La expresión genética se denomina como dominante o recesiva, también ambos alelos de un par de genes pueden expresar completamente en el heterocigoto y se denomina codominancia como se ve con los antígenos de histocompatibilidad y grupos sanguíneos. Un gen mutante único da lugar a efectos finales, que se denomina pleiotropismo. 11

12
PATRONES DE TRANSMISIÓN DE LAS ENFERMEDADES MONOGÉNICAS
Mutaciones que afectan genes únicos siguen tres patrones de herencia Autosómico Dominante Autosómico Recesivo Ligado al Cromosoma X

13
ENFERMEDADES MENDELIANAS
Enfermedades Autosómico Dominantes Heterocigoto. Alguno de los padres esta afectado No hay distinción de genero Hijo de persona afectada y persona no afectada. 50% de posibilidades

14
Enfermedades Autosómicas Dominantes
CARACTERISTICAS Algunos pacientes no tienen padres afectados. En ellos se deben a mutaciones de novo en células germinales (óvulo o espermatozoide). Padres mayores, drogas etc. Los signos clínicos pueden modificarse por una penetrancia reducida y una expresividad variable. Algunos individuos heredan el gen mutante pero son fenotípicamente normales. En muchas enfermedades la edad de inicio está retrasada, o sea que los síntomas y signos no aparecen hasta la edad adulta como en la enfermedad de Huntington
. Padres mayores, drogas etc. Los signos clínicos pueden modificarse por una penetrancia reducida y una expresividad variable. Algunos individuos heredan el gen mutante pero son fenotípicamente normales. En muchas enfermedades la edad de inicio está retrasada, o sea que los síntomas y signos no aparecen hasta la edad adulta como en la enfermedad de Huntington.")
15
Enfermedades Autosómico Dominantes
Nervioso Esquelético Urinario Hematopoyético Neurofibromatosis Síndrome de Marfan Enfermedad renal poliquistica Esferocitosis hereditaria Distrofia Miotónica Síndrome de Ehlers-Darlos Enfermedad de Von Willebrand Esclerosis tuberosa Osteogenesis imperfecta Acondroplasia

17
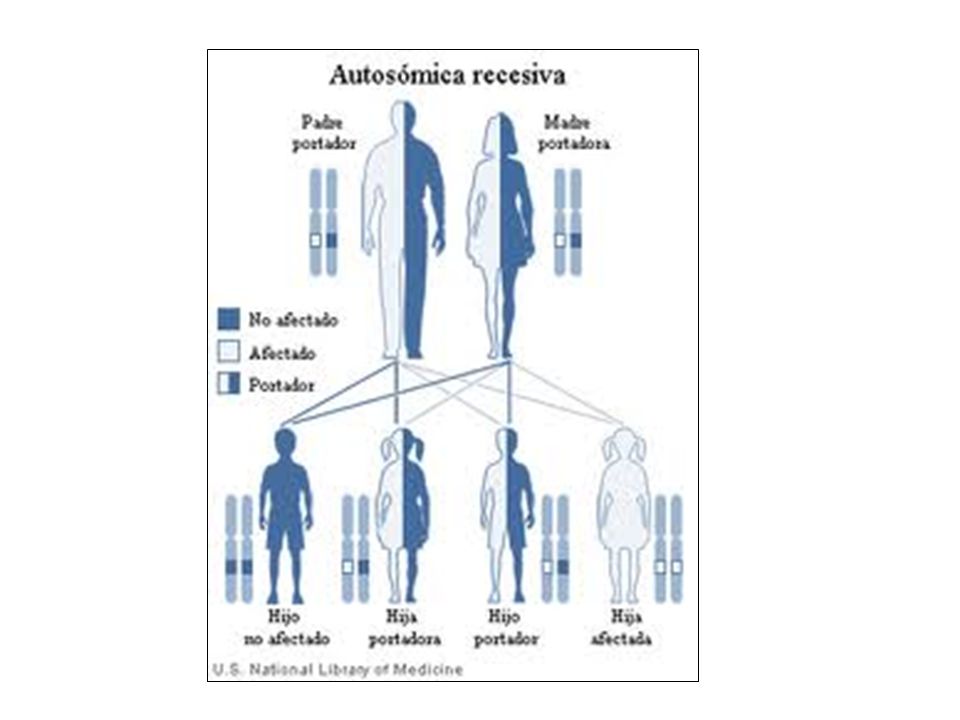
ENFERMEDADES AUTOSÓMICAS RECESIVAS
La categoría de trastornos mendelianos más grande. Se produce cuando ambos alelos de un determinado locus génico son mutantes, por lo que se caracterizan por los siguientes datos: no suele afectar a los padres, pero los hermanos pueden tener la enfermedad. los hermanos tienen una posibilidad sobre cuatro de estar afectados (es decir 25% para cada nacimiento). si el gen mutante ocurre con baja frecuencia en la población, existe una fuerte probabilidad de que el sujeto sea el producto de un matrimonio consanguíneo. 17
. si el gen mutante ocurre con baja frecuencia en la población, existe una fuerte probabilidad de que el sujeto sea el producto de un matrimonio consanguíneo. 17.")
18
ENFERMEDADES AUTOSÓMICAS RECESIVAS
La expresión del defecto tiende a ser más uniforme que en las dominantes B. La penetrancia completa es frecuente. C. El inicio suele presentarse precozmente en la vida. Ocurren mutaciones nuevas por trastornos recesivos que muy pocas veces se detectan clínicamente. En muchos casos las proteínas enzimáticas están afectadas por una pérdida de función. 18

20
Enfermedades Autosómicos Recesivos
Nervioso Metabólico Endocrino Hemtopoyetico Esqueletico Atrofia musculares neurogenicas Fibrosis Quística Hiperplasia suprarrenal congénita Anemia de células falciformes Síndrome de Ehlers-Danlos Ataxia de Friedreich Fenilcetonuria Talasemias Alcaptonuria Atrofia muscular especial Galactosemia Hemocromatosis

21
ENFERMEDADES LIGADAS AL CROMOSOMA X.
Todas las enfermedades ligadas al sexo son enfermedades ligadas al cromosoma X. Casi todas las ligadas al cromosoma X son recesivas. El cromosoma Y de los varones sufre mutaciones pero los pacientes son infértiles y por lo tanto no existe herencia ligada al cromosoma Y. 21

22
Enfermedades ligadas al Cromosoma X
Nervioso Esquelético Hematopoyético Metabólico Inmunidad Síndrome del Cromosoma X frágil Distrofia Muscular de Duchenne Hemofilia A Diabetes insipida Agammaglobulinemia Hemofilia B Síndrome de Lesch-Nyhan Síndrome de Wiskott-Aldrich

23
CROMOSOMA X FRAGIL FraxA (Xq27,3-28)
Sitio frágil cerca del telómero FRAX A Xq27,3 FRAX E Xq28 Repeticiones de triplete deja sitio frágil Primera causa de retraso mental hereditario

24
ENFERMEDADES ASOCIADAS CON DEFECTOS EN PROTEÍNAS ESTRUCTURALES.
SINDROME DE MARFAN SINDROME DE EHLERS-DANLOS 24

25
SINDROME DE MARFAN Es un trastorno de los tejido conectivos del organismo, y se manifiesta principalmente por cambios en el esqueleto, los ojos y el sistema cardiovascular. Prevalencia 1/5000 70 al 85% son familiares, herencia autosómica dominante, resto por mutaciones nuevas. 25

27
SINDROME DE MARFAN Defecto hereditario en una glucoproteína extracelular llamada fibrilina-1. Fibrilina-1 es el componente de las microfibrillas de la matriz extracelular y llegan a formar las fibras elásticas. Las Fibras elásticas : aorta, ligamentos y zónulas ciliares del cristalino. La fibrilina tiene dos formas: 1 y 2, que están codificadas en 2 genes FBN1 y FBN2. Las mutaciones del FBN1 dan lugar al síndrome de Marfan. Las mutaciones del FBN2 dan lugar a la aracnodactilia contractural congénita (anomalías esqueléticas) 27
 27.")
28
SÍNDROME DE MARFAN Morfología: anomalías esqueléticas
Excesivamente alto, de extremidades largas, con dedos largos en manos y pies. Cambios oculares los característicos son las subluxación o luxación (hacia fuera y arriba) bilateral del cristalino (ectopia lentis). Lesiones cardiovasculares: prolapso de la válvula mitral (de mayor importancia), y la dilatación de la aorta ascendente con necrosis quistica de la media. 28
 bilateral del cristalino (ectopia lentis). Lesiones cardiovasculares: prolapso de la válvula mitral (de mayor importancia), y la dilatación de la aorta ascendente con necrosis quistica de la media. 28.")
30
Desordenes Citogeneticos
CLASIFICACION DE ENFERMEDADES GENETICAS ALTERACIONES CROMOSOMICAS ALTERACIONES DE GENES SIMPLES ALTERACIONES MULTIFACTORIALES

31
Desordenes Citogeneticos
ANOMALIAS NUMERICAS Aneuploidías (monosomías, trisomías, tetrasomías) Poliploidías (triploidía, tetraploidía) ANOMALIAS ESTRCUTURALES traslocaciones recíprocas y roberthsonianas Deleciones, microdelecciones inserciones inversiones Cromosomas en anillo
 Poliploidías (triploidía, tetraploidía) ANOMALIAS ESTRCUTURALES. traslocaciones recíprocas y roberthsonianas. Deleciones, microdelecciones. inserciones. inversiones. Cromosomas en anillo.")
32
Desordenes Citogeneticos
SINDROMES SINDROME DE DOWN SINDROME DE EDWARDS SINDROME DE PATAU SINDROME DE KLINEFELTER SINDROME DE TURNER

33
Síndrome de Down 15% transmitido por espermatozoide
85% transmitido por el óvulo La expresión bioquímica del síndrome consiste en el aumento de diferentes enzimas. Exceso de superoxido dismutasa: se acumula H2O2, peroxidación de lípidos, proteínas y DNA

34
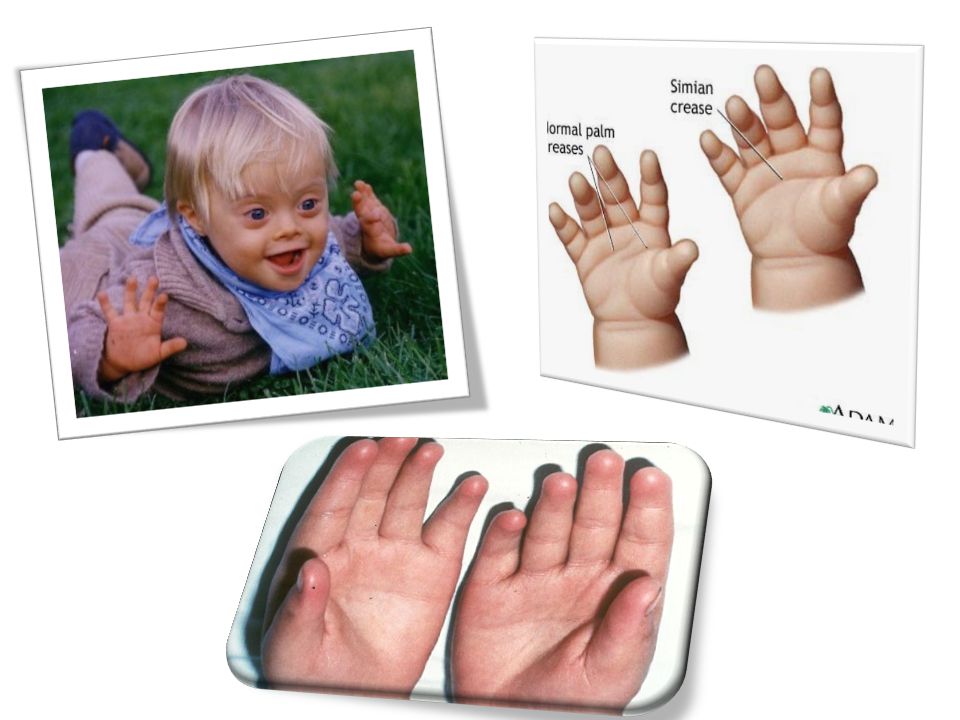
SÍNDROME DE DOWN 1/600- 1/800 RNV Malformaciones cardiacas
Retraso mental (C.I. 60) Hipotonía e hiperflexibilidad muscular Fascies de perfil achatado Ángulos palpebrales externos inclinados hacia arriba Pliegue en epicanto interno
 Hipotonía e hiperflexibilidad muscular. Fascies de perfil achatado. Ángulos palpebrales externos inclinados hacia arriba. Pliegue en epicanto interno.")
36
SINDROME DE DE DOWN Madre 25 años 1 cada 2000 RNV
Madre de 35 años 1 cada 200 RNV Madre de más de 40 años 1 cada 40 RNV

37
SINDROME DE DOWN Esperanza de vida 40 o 60 años
Malformaciones cardiacas o leucemias causa de muerte prematura Paladar ojival 70% Hipodoncia 60% Macroglosia 43% Cardiopatías congénitas 45%

38
SÍNDROME DE EDWARDS 1/3000 - 1/7000 RNV Proporción 1:3, niños : niñas
Sobrevida 10 años Micrognatia, paladar ojival, fisura labial y palatina, hipertelolismo, microftlamia Manos: dedo índice abraza al pulgar y anular y meñique al medio Malformaciones renales y cardiacas

39
TRISOMÍA 18 (SÍNDROME DE EDWARDS)
")
40
TRISOMÍA CROMOSOMA 18

41
SÍNDROME DE PATAU 1/10.000- 1/20.000 Sobrevida 1 año
No hay diferencias por sexo Cráneo con escasa calcificación, microcefalia, anoftalmia, microftamia, cataratas, nariz aplanada y grande, orejas displásicas y baja implantación. Fisuras labiales y palatinas

42
TRISOMÍA 13(47,XX+13)
")
43
SINDROME DE KLINEFELTER
Incidencia aproximadamente 1:700 recién nacidos vivos varones Gran variación clínica Algunos con apariencia normal Estériles Nivel intelectual normal o déficit verbal Hipogonadismo y déficit andrógenos

44
SÍNDROME KLINEFELTER(47,XXY) trisomía
 trisomía")
45
CARIOTIPO TRISOMÍA 21 (47,XX+21 o 47, XY +21)
")
46
SINDROME DE TURNER El diagnóstico en el momento del nacimiento se da por la presencia de un edema o acumulación excesiva de líquido seroalbuminoso en el tejido celular característico en el dorso de manos y pies, así como por los pliegues laxos de piel en la nuca. Son frecuentes bajo peso y talla corta al nacer. Una de cada mujeres nacidas en sufre el Síndrome de Turner (ST)
")
47
SÍNDROME DE TURNER(45,X) Descrito por primera vez en 1938, su origen cromosómico no se descubrió hasta 1959.
 Descrito por primera vez en 1938, su origen cromosómico no se descubrió hasta")
48
SINDROME DE TURNER Problemas de crecimiento
Malformaciones del corazón 75% Malformaciones renales (en herradura) Alteraciones del sistema linfático Malformaciones de los oídos asociados a pérdidas auditivas. 75% otitis Infértiles, propensas a osteoporosis
 Alteraciones del sistema linfático. Malformaciones de los oídos asociados a pérdidas auditivas. 75% otitis. Infértiles, propensas a osteoporosis.")
49
SINDROME DE TURNER Línea más baja de implantación posterior del cabello Mandíbula pequeña Orejas prominentes Convexidad excesiva de las uñas, lechos ungueales pequeños

50
SÍNDROME DE TURNER Monosomía: 45, X0

Presentaciones similares