Descargar la presentación
La descarga está en progreso. Por favor, espere

1
Farmacocinética en el paciente crítico
Luis Enríquez, MD. Residente Anestesiología Tutores: Dr. O. Medina y Dr. O. Cañas Diciembre, 2011

2
Introducción El paciente crítico tiene un espectro de disfunción orgánica Se requiere tratamiento con una variedad de medicamentos Para individualizar cada paciente es necesario conocer algunos principios generales de los procesos que se afectan en los pacientes críticos Critically ill patients exhibit a range of organ dysfunctions and often require treatment with a variety of drugs including sedatives, analgesics, neuromuscular blockers, antimicrobials, inotropes and gastric acid suppressants. Understanding how organ dysfunction can alter the pharmacokinetics of drugs is a vital aspect of therapy in this patient group. Additional factors that may impact on drug pharmacokinetics include drug interactions (e.g. warfarin and aminophylline) and other therapeutic interventions (e.g. dialysis).
 and other therapeutic. interventions (e.g. dialysis).")
3
Manejo del paciente crítico
Soporte general de los órganos en falla Tratamiento especifico del proceso de base Prevención de complicaciones Lograr una concentración efectiva y segura de medicamentos depende de factores del medicamento y de factores del paciente The management of critically ill patients encompasses: (i) specific treatment of the disease process; (ii) general support of failing organs during natural healing and repair, including nutrition and maintenance of fluid and electrolyte balance; and finally, (iii) the avoidance and treatment of complications (e.g. sepsis). Many of these processes require the administration of drugs. The ultimate aim of drug administration in the critically ill patient is to achieve a safe and effective concentration of the drug in the target tissue.[1,2] The ability to achieve this aim will depend on drug-related factors, such as the dose administered and disposition characteristics, as well as patient-specific factors such as drug delivery to the site of action (e.g. cardiac output).
 specific treatment of the disease. process; (ii) general support of failing organs during. natural healing and repair, including nutrition. and maintenance of fluid and electrolyte balance; and finally, (iii) the avoidance and treatment of. complications (e.g. sepsis). Many of these processes. require the administration of drugs. The ultimate. aim of drug administration in the critically. ill patient is to achieve a safe and effective concentration. of the drug in the target tissue.[1,2] The ability. to achieve this aim will depend on drug-related. factors, such as the dose administered and disposition. characteristics, as well as patient-specific factors. such as drug delivery to the site of action. (e.g. cardiac output).")
4
Definiciones Farmacocinética Farmacodinamia
Estudio de los procesos que determinan la concentración de un medicamento en su sitio de acción Que le hace el cuerpo al medicamento Estudio de los procesos bioquímicos y fisiológicos de los medicamentos, mecanismos de acción y relación entre concentración y efecto Que le hace el medicamento al cuerpo

5
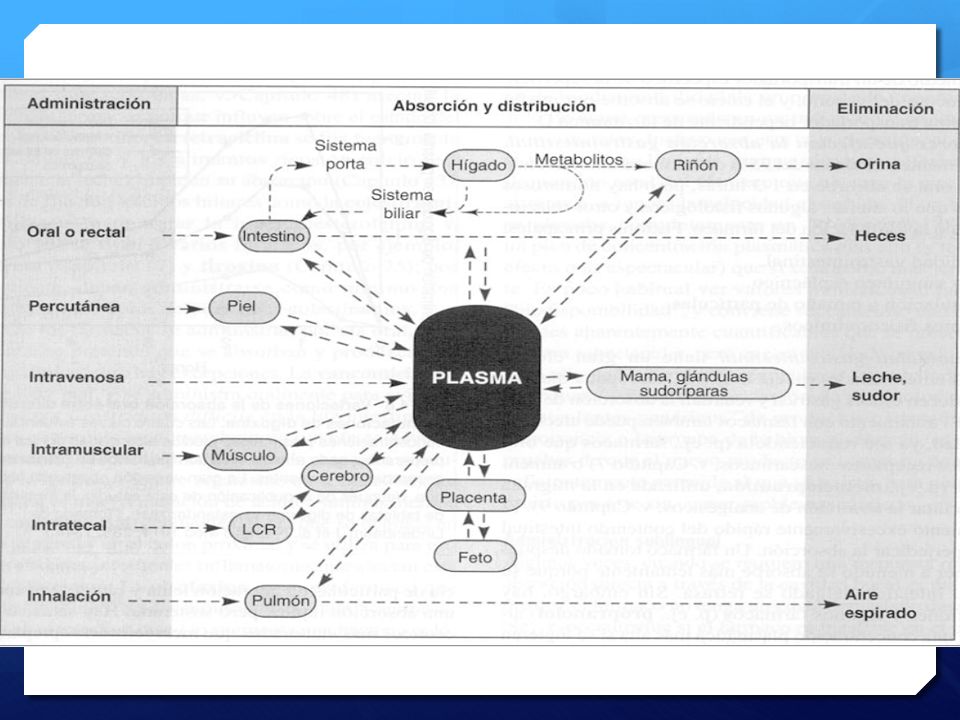
Mecanísmos farmacocinéticos básicos
Metabolísmo Excreción de metabolítos Absorción del medicamento Excreción del medicamento Transporte en sangre Absorción : Movimiento de un fármaco desde el sitio de administración hasta la circulación sanguínea. : Distribución : Proceso por el que un fármaco difunde o es transportado desde el espacio intravascular hasta los tejidos y células corporales. Metabolismo : Conversión química o transformación, de fármacos o sustancias endógenas, en compuestos más fáciles de eliminar. Eliminación : Excreción de un compuesto, metabolito o fármaco no cambiado, del cuerpo mediante un proceso renal, biliar o pulmonar. Distribución en los tejidos Boucher, B. et al. Pharmacokinetic Changes in Critical Illness, Crit Care Clin 22 (2006) 255– 271
 255– 271.")
8
Conceptos básicos

9
Absorción en el paciente crítico
Farmaco: Tamaño, solubilidad, lipofilicidad, pKa, estabilidad Entorno: pH, flujo sanguíneo, área de superficie, motilidad GI Vía de elección: I.V para la mayoría de los medicamentos patient (eg, IV esmolol rather than oral long-acting metoprolol preparations for rate control of supraventricular tachycardia [SVT] in a patient who has congestive heart failure [CHF]). size, solubility, degree of lipophilicity, pKa, and stability are important factors influencing the rate and extent of drug absorption. Environmental characteristics that can affect drug absorption include pH, blood flow, surface area, and gastrointestinal (GI) motility. Inicio – Predictibilidad - Titulabilidad
. size, solubility, degree of lipophilicity, pKa, and stability are important factors. influencing the rate and extent of drug absorption. Environmental characteristics. that can affect drug absorption include pH, blood flow, surface area, and. gastrointestinal (GI) motility. Inicio – Predictibilidad - Titulabilidad.")
10
Enoxaparina Crit Care Med 2003 Vol. 31, No. 5 1405
Background: Subcutaneously administered low-molecularweight heparins are widely used for prevention of venous thromboembolism. The appropriateness of the subcutaneous route in critically ill patients has never been established. Objective: To determine anti-Xa activities in critically ill patients and in noncritically ill patients receiving prophylactic doses of subcutaneous enoxaparin. Design: Prospective, controlled, open-labeled study. Setting: Tertiary medical-cardiologic-postoperative intensive care unit and a general medical ward at a university hospital. Patients: A total of 16 intensive care unit patients (group 1; age, yrs; male/female ratio, 7/9; Acute Physiology and Chronic Health Evaluation II score, ; mechanical ventilation, n 15; vasopressors, n 13) and 13 noncritically ill medical patients (group 2; age, yrs; male/female ratio, 7/6) were studied. Body mass index ( vs kg/m2, p not significant) was comparable and serum creatinine levels ( vs mg/dL, group 1 vs. 2) were within the normal range in both groups. Patients with impaired renal function, receiving hemofiltration, or requiring therapeutic anticoagulation were not eligible. Interventions: None. Measurements and Main Results: Anti-Xa activities were determined at 0, 1, 3, 6, and 12 hrs after a single daily subcutaneous dose of 40 mg enoxaparin on day 1 and at 3 hrs after 40 mg of enoxaparin on days 2–5. Mean anti-Xa levels at 0 to 12 hrs were consistently lower in group 1 compared with group 2 by analysis of variance (p between groups and over time), as was the area under the curve at 0 to 12 hrs ( vs units·mL1·hr1, group 1 vs. 2, p .008). Significant differences in anti-Xa activity were also found on days 2–5 (p .001). Peak anti-Xa activities at 3 hrs after administration were negatively correlated with the body mass index (r .41, p < .03). No correlation was found between the anti-Xa activity at 3 hrs and the dose of norepinephrine (r .12, p .7). Conclusion: Critically ill patients with normal renal function demonstrated significantly lower anti-Xa levels in response to a single daily dose of subcutaneous enoxaparin when compared with medical patients in the normal ward. (Crit Care Med 2003; 31:1405–1409) KEY WORDS: enoxaparin; anti-Xa activity; critically ill; intensive Crit Care Med 2003 Vol. 31, No
 and 13 noncritically ill. medical patients (group 2; age, yrs; male/female ratio, 7/6) were studied. Body mass index ( vs kg/m2, p not significant) was comparable and serum creatinine levels. ( vs mg/dL, group 1 vs. 2) were within the. normal range in both groups. Patients with impaired renal function, receiving hemofiltration, or requiring therapeutic anticoagulation. were not eligible. Interventions: None. Measurements and Main Results: Anti-Xa activities were determined. at 0, 1, 3, 6, and 12 hrs after a single daily subcutaneous. dose of 40 mg enoxaparin on day 1 and at 3 hrs after 40 mg of. enoxaparin on days 2–5. Mean anti-Xa levels at 0 to 12 hrs were. consistently lower in group 1 compared with group 2 by analysis. of variance (p .001 between groups and over time), as was the. area under the curve at 0 to 12 hrs (2.6 1 vs units·mL1·hr1, group 1 vs. 2, p .008). Significant differences. in anti-Xa activity were also found on days 2–5 (p .001). Peak. anti-Xa activities at 3 hrs after administration were negatively. correlated with the body mass index (r .41, p < .03). No. correlation was found between the anti-Xa activity at 3 hrs and. the dose of norepinephrine (r .12, p .7). Conclusion: Critically ill patients with normal renal function. demonstrated significantly lower anti-Xa levels in response to. a single daily dose of subcutaneous enoxaparin when compared. with medical patients in the normal ward. (Crit Care Med. 2003; 31:1405–1409) KEY WORDS: enoxaparin; anti-Xa activity; critically ill; intensive. Crit Care Med 2003 Vol. 31, No")
11
Absorción enteral Para medicamentos que se absorben entre el estomago y el cólon puede existir metabolísmo de primer paso Flora enzimas intestinales, hepáticas, pancreáticas pueden inactivar medicamentos (morfina, propranolol) ó activarlos (Enalapril, Enalaprilat) Para conversión de ciclosporina oral a IV: dosis por 1/3 Varía si se administran otros medicamentos como Itraconazol
 ó activarlos (Enalapril, Enalaprilat) Para conversión de ciclosporina oral a IV: dosis por 1/3. Varía si se administran otros medicamentos como Itraconazol.")
12
Reducida absorción intestinal
Atrofia intestinal Hipomotilidad Vellosidades disminuidas Función celular comprometida Función enzimática comprobada disminuida Posible alteración en absorción Por ileo, falta de estímulo, medicamentos Especialmente opioides Puede comprometer vaciamiento gástrico Posible alteración en absorción Tambien el edema intestinal

13
Alteraciones en función de barrera
Paciente con sepsis Efecto de citoquinas y efecto citopático de endotoxina Causa un área menor de absorción Incremento relativo de poros grandes Posibles alteraciones secundarias en absorción

14
Incompatibilidad física
Absorción Incompatibilidad física Alteraciones en pH intestinal y su influencia en el estado de ionización de los medicamentos Ejemplo: Requerimento de pH ácido para absorción de Itraconazol vs Profilaxis enfermedad ácido péptica Otro problema es la administración concurrente con nutrición enteral La warfarina se une a la formula Evidencia in vitro. El PT se prolonga cuando se suspende la vía enteral Fenitoina, minociclina y tetraciclina: Evidencia clínica Datos inconcluyentes para fluconazol y ciprofloxacina Frequent changes in pH occur in the critically ill patient as a result of numerous conditions, such as respiratory failure, shock states, and renal failure. As previously mentioned, the pH of the environment affects the ionized state of many drugs. It is well understood that the ionized drug does not penetrate the lipid-based cellular membrane as easily. Therefore, alterations in the ionized state can increase or decrease the extent of distribution of a drug. Because pH changes accompany many other physiologic alterations in critical illness, it is difficult to isolate the degree of impact that pH changes have on distribution. As a result, direct evidence of such effects is limited.

15
Distribución La distribución depende de varios factores, incluyendo:
Gasto cardiaco, unión a proteinas, permeabilidad de los tejidos, liposolubilidad, pH del ambiente, pKa del medicamento. Modelos complejos para incorporar estas variables, sin embargo el modelo bicompatimental funciona bien para la mayoría de los medicamentos Using the most simple pharmacokinetic model, a one-compartment model, distribution of a drug can be mathematically represented by the equation C=D/ Vd, where C is the initial concentration of a drug administered as an intravenous bolus, D is the dose, and Vd is the volume of distribution. Distribution of most drugs to the various bodily tissues is dependent on multiple factors, such as blood delivery, degree of protein binding, permeability of the tissues, lipid solubility of the drug, pH of the environment, and pKa of the drug, however. Incorporating these complex interactions requires more intricate pharmacokinetic modeling necessitating the assistance of computers. Surprisingly, a simplified twocompartment model similar to Fig. 1 works well for most drugs. During critical

16
Aplicación clínica del VD
Distribución Aplicación clínica del VD Dosis de carga C=D/Vd C x Vd=D

17
Concentración objetivo
Distribución Concentración objetivo Regimen empírico Guiado por antibiograma El antibiograma reporta la MIC Se pueden medir concentraciones pico y valle Tanto subdosificación como sobredosificación Previene falta de efecto y toxicidad

18
Farmacocinética de acuerdo a parametros farmacodinámicos
Distribución Farmacocinética de acuerdo a parametros farmacodinámicos Concentración pico blanco para aminoglucosidos: 8 a 10 veces la MIC Area sobre la MIC: 100mgxh/L en 24 horas Regimenes de ¨dosis de intervalos extendidos¨ Menos nefrotoxicidad igual ototoxicidad Efecto postantibiotico Aminoglycosides constitute one of the oldest classes of antimicrobials. Despite their toxicity, mainly nephrotoxicity and ototoxicity, aminoglycosides are valuable in current clinical practice, since they retain good activity against multidrug-resistant Gram-negative pathogens, such as Pseudomonas aeruginosa and Acinetobacter spp. Time-kill studies have shown a concentration-dependent and partially concentration-dependent bacterial killing against Gram-negative and Gram-positive bacteria, respectively. Pharmacodynamic data gathered over recent decades show that the administration of aminoglycosides by an extended-interval dosing scheme takes advantage of the maximum potential of these agents, with the goal of achieving an area under the concentration-time curve (AUC) of 100 mg · h/L over 24 hours and a peak plasma drug concentration (Cmax) to minimum inhibitory concentration (MIC) ratio of 8–10. Several clinical conditions that are common in seriously ill patients result in expansion of the extracellular space and can lead to a lower than desirable Cmax with the usual loading dose. Extended-interval dosing schemes allow adequate time to decrease bacterial adaptive resistance, a phenomenon characterized by slow concentration-independent killing. Adaptive resistance is minimized by the complete clearance of the drug before the subsequent dose, thus favouring the extended-interval dosing schemes. The efficacy of these schemes is also safeguarded by the observed post-antibiotic sub-MIC effect and post-antibiotic leukocyte enhancement, which inhibit bacterial regrowth when the serum aminoglycoside levels fall below the MIC of the pathogen. In everyday clinical practice, aminoglycosides are usually used empirically to treat severe sepsis and septic shock while awaiting the results of antimicrobial susceptibility testing. The European Committee on Antimicrobial Susceptibility Testing acknowledges the regimen-dependent nature of clinical breakpoints for aminoglycosides, i.e. of MIC values that classify bacterial isolates into sensitive or resistant, and bases its recommendations on extended-interval dosing. To a large extent, the lack of correlation between in vitro antimicrobial susceptibility testing and clinical outcome is derived from the fact that the available clinical breakpoints for aminoglycosides are set based on mean pharmacokinetic parameters obtained in healthy volunteers and not sick patients. The nephrotoxicity associated with once- versus multiple-daily administration of aminoglycosides has been assessed in numerous prospective randomized trials and by several meta-analyses. The once-daily dosing schedule provides a longer time of administration until the threshold for nephrotoxicity is met. Regarding ototoxicity, no dosing regimen appears to be less ototoxic than another. Inactivation of aminoglycosides inside the bacterial pathogens occurs by diverse modifying enzymes and by operation of multidrug efflux systems, making both of these potential targets for inhibition. In summary, despite their use for several decades, the ideal method of administration and the preferred dosing schemes of aminoglycosides for most of their therapeutic indications need further refinement. Individualized pharmacodynamic monitoring has the potential of minimizing the toxicity and the clinical failures of these agents in critically ill patients. Pagkalis, S. et al. Pharmacological Considerations for the Proper Clinical Use of Aminoglycosides. DRUGS Volume 71 - Issue 17 - pp
 of 100 mg · h/L over 24 hours and a peak plasma drug concentration (Cmax) to minimum inhibitory concentration (MIC) ratio of 8–10. Several clinical conditions that are common in seriously ill patients result in expansion of the extracellular space and can lead to a lower than desirable Cmax with the usual loading dose. Extended-interval dosing schemes allow adequate time to decrease bacterial adaptive resistance, a phenomenon characterized by slow concentration-independent killing. Adaptive resistance is minimized by the complete clearance of the drug before the subsequent dose, thus favouring the extended-interval dosing schemes. The efficacy of these schemes is also safeguarded by the observed post-antibiotic sub-MIC effect and post-antibiotic leukocyte enhancement, which inhibit bacterial regrowth when the serum aminoglycoside levels fall below the MIC of the pathogen. In everyday clinical practice, aminoglycosides are usually used empirically to treat severe sepsis and septic shock while awaiting the results of antimicrobial susceptibility testing. The European Committee on Antimicrobial Susceptibility Testing acknowledges the regimen-dependent nature of clinical breakpoints for aminoglycosides, i.e. of MIC values that classify bacterial isolates into sensitive or resistant, and bases its recommendations on extended-interval dosing. To a large extent, the lack of correlation between in vitro antimicrobial susceptibility testing and clinical outcome is derived from the fact that the available clinical breakpoints for aminoglycosides are set based on mean pharmacokinetic parameters obtained in healthy volunteers and not sick patients. The nephrotoxicity associated with once- versus multiple-daily administration of aminoglycosides has been assessed in numerous prospective randomized trials and by several meta-analyses. The once-daily dosing schedule provides a longer time of administration until the threshold for nephrotoxicity is met. Regarding ototoxicity, no dosing regimen appears to be less ototoxic than another. Inactivation of aminoglycosides inside the bacterial pathogens occurs by diverse modifying enzymes and by operation of multidrug efflux systems, making both of these potential targets for inhibition. In summary, despite their use for several decades, the ideal method of administration and the preferred dosing schemes of aminoglycosides for most of their therapeutic indications need further refinement. Individualized pharmacodynamic monitoring has the potential of minimizing the toxicity and the clinical failures of these agents in critically ill patients. year=2011&issue=71170&article=00004&type=abstract. Pagkalis, S. et al. Pharmacological Considerations for the Proper Clinical Use of Aminoglycosides. DRUGS Volume 71 - Issue 17 - pp")
19
Aplicación: Problema con los Macrólidos
Distribución Aplicación: Problema con los Macrólidos Adulto joven, sepsis por Pseudomona, 1.65cm estatura, peso 80kg al ingreso y 100kg luego de reanimación. IBW: 60kg VD trobamicina: 0.25 L/kg Concentración blanco: 8mg/L DOSIS DE CARGA: 60kg (0.25L/kg)(8mg/L)= 120mg DOSIS por peso= 1.5 a 2 mg/kg IBW= 90 a 120 mg Ejercicio trobamicina: body weight (IBW) can be calculated from the patient’s height: Men: IBW <eth>kg? 1/4 50 ? <eth>2:3 _1/2height in inches greater than 5 feet_? <eth>3? Women: IBW <eth>kg? 1/4 50 ? <eth>2:3 greater than 5 feet_? <eth>4? Adjustments to the total Vd can be made according to certain clinical features. For obese patients, the additional weight above the IBW may affect the total Vd. For lipophilic drugs, such as many sedatives, the entire excess body weight should be included in the calculation of loading dose; for hydrophilic drugs, such as aminoglycosides, a factor of 10% x (ABW _ IBW) should be added to the Vd to account for the water in the excess weight compartment. Critical illness can rapidly change total body weight, particularly when septic shock requires multiple liters of volume resuscitation, and the additional fluid weight increases the Vd of hydrophilic drugs. For hydrophilic drugs, the net increase in body weight secondary to fluid resuscitation should be added to the total Vd. Let us consider an example to further illustrate this point. A man who has pseudomonas sepsis requires aminoglycoside therapy. He is 64 inches tall, weighs 80 kg on admission, and after fluid resuscitation, weights 100 kg. By using Equation 3, we can calculate his IBW to be approximately 60 kg. Given that the Vd of tobramycin is 0.25 L/kg and the desired plasma concentration of tobramycin is 8 mg/L, we can calculate theLD based on IBWusing Equation 2 to be: 60 kg (0.25 L/kg) (8 mg/L) = 120 mg. This is approximately the same loading dose that would be prescribed using standard weight-based dosing (1.5–2 mg/kg LBW). However, neither the use of standard Vd data nor the use of mg/kg dosing will provide an adequate loading dose in this patient; 120 mg does not come close to achieving the target plasma concentration. vs
(8mg/L)= 120mg. DOSIS por peso= 1.5 a 2 mg/kg IBW= 90 a 120 mg. Ejercicio trobamicina: body weight (IBW) can be calculated from the patient’s. height: Men: IBW <eth>kg 1/4 50 <eth>2:3. _1/2height in inches. greater than 5 feet_ <eth>3 Women: IBW <eth>kg 1/4 50 <eth>2:3. greater than 5 feet_ <eth>4 Adjustments to the total Vd can be made according. to certain clinical features. For obese patients, the. additional weight above the IBW may affect the total. Vd. For lipophilic drugs, such as many sedatives, the. entire excess body weight should be included in the. calculation of loading dose; for hydrophilic drugs, such as aminoglycosides, a factor of 10% x (ABW _. IBW) should be added to the Vd to account for the. water in the excess weight compartment. Critical. illness can rapidly change total body weight, particularly. when septic shock requires multiple liters of. volume resuscitation, and the additional fluid weight. increases the Vd of hydrophilic drugs. For hydrophilic. drugs, the net increase in body weight secondary. to fluid resuscitation should be added to the. total Vd. Let us consider an example to further illustrate this. point. A man who has pseudomonas sepsis requires. aminoglycoside therapy. He is 64 inches tall, weighs. 80 kg on admission, and after fluid resuscitation, weights 100 kg. By using Equation 3, we can calculate. his IBW to be approximately 60 kg. Given that the Vd. of tobramycin is 0.25 L/kg and the desired plasma. concentration of tobramycin is 8 mg/L, we can calculate. theLD based on IBWusing Equation 2 to be: 60 kg. (0.25 L/kg) (8 mg/L) = 120 mg. This is approximately the same loading dose that. would be prescribed using standard weight-based. dosing (1.5–2 mg/kg LBW). However, neither the. use of standard Vd data nor the use of mg/kg dosing. will provide an adequate loading dose in this patient; 120 mg does not come close to achieving the target. plasma concentration. vs.")
20
Distribución Definitivamente, 20L en reanimación cambian
la farmacocinética…. Rea RS, Capitano B. Optimizing use of aminoglycosides in the critically ill. Semin Respir Crit Care Med Dec;28(6): Review. Mann HJ, et al. Altered aminoglycoside pharmacokinetics in critically ill patients with sepsis.Clin Pharm. 1987 Feb;6(2): Pagkalis, S. et al. Pharmacological Considerations for the Proper Clinical Use of Aminoglycosides. DRUGS Volume 71 - Issue 17 - pp
: Review. Mann HJ, et al. Altered aminoglycoside pharmacokinetics in critically ill patients with sepsis.Clin Pharm Feb;6(2): Pagkalis, S. et al. Pharmacological Considerations for the Proper Clinical Use of Aminoglycosides. DRUGS Volume 71 - Issue 17 - pp")
21
Efecto del aumento del VD
Distribución Efecto del aumento del VD Macrolidos: Hidrofílicos VD influenciado por componente hidrofílico en tejido adiposo + “Tercer espacio” El VD se puede considerar: VD usando el IBW + Componente hídrico del tejido adiposo: 10% del exceso en Kg Exceso en peso por tercer espacio en Kg = (0.25 L/kg x 60 Kg) + (0.10 x 20kg) + (100 kg – 80 kg) = 37L Because tobramycin is a hydrophilic drug, its Vd is influenced by the additional hydrophilic component in the excess adipose tissue, as well as the extra third-spaced volume following resuscitation from shock. A more accurate calculation of Vd would include the following: <eth>Vd using IBW? ? <eth>10% of excess adipose weight; in kg? ?<eth>excess third-spacing weight; in kg? 1/4 <eth>0:25 L=kg _ 60 kg? ? <eth>0:10 _ 20 kg? ?<eth>100 kg _ 80 kg? 1/4 37L The corresponding calculation of loading dose using the adjusted Vd is 37 L _ 8 mg/L = 296 mg. If the volume of distribution is not adjusted for the massive increases that are observed with sepsis and (to a lesser extent) obesity, then the calculated loading dose would be grossly underestimated and the observed peak plasma concentration of aminoglycoside would be inadequate for optimal concentration-dependent bacterial killing at the time of greatest urgency. Studies indicate that loading doses of aminoglycosides are routinely underestimated using standard dosing strategies; adjustments for increased Vd are crucial to successful initiation of therapy in this setting 2
 + (0.10 x 20kg) + (100 kg – 80 kg) = 37L. Because tobramycin is a hydrophilic. drug, its Vd is influenced by the additional. hydrophilic component in the excess adipose tissue, as well as the extra third-spaced volume following. resuscitation from shock. A more accurate calculation. of Vd would include the following: <eth>Vd using IBW <eth>10% of excess adipose weight; in kg <eth>excess third-spacing weight; in kg 1/4 <eth>0:25 L=kg _ 60 kg <eth>0:10 _ 20 kg <eth>100 kg _ 80 kg 1/4 37L. The corresponding calculation of loading dose. using the adjusted Vd is 37 L _ 8 mg/L = 296 mg. If the volume of distribution is not adjusted for the. massive increases that are observed with sepsis and (to. a lesser extent) obesity, then the calculated loading. dose would be grossly underestimated and the observed. peak plasma concentration of aminoglycoside. would be inadequate for optimal concentration-dependent. bacterial killing at the time of greatest urgency. Studies indicate that loading doses of aminoglycosides. are routinely underestimated using standard dosing. strategies; adjustments for increased Vd are crucial. to successful initiation of therapy in this setting. 2.")
22
Dosis de carga ajustada al nuevo VD
Distribución Dosis de carga ajustada al nuevo VD VD x C = D 37 L x 8 mg/L = 296 mg DOSIS DE CARGA con VD no ajustado: 60kg (0.25L/kg)(8mg/L)= 120mg DOSIS por peso= 1.5 a 2 mg/kg IBW= 90 a 120 mg
(8mg/L)= 120mg. DOSIS por peso= 1.5 a 2 mg/kg IBW= 90 a 120 mg.")
23
Obtención de datos sobre medicamentos
Micromedex™ Cerner Multum™ Wolters Kluwer™

24
Ejemplos de VD Medicamento VD Comentario Warfarina 8L
Alta unión a proteinas plasmáticas Teofilina, Etanol 30L Distribución en el agua corporal total Cloroquina 15000L Alta liposolubilidad, secuestro en tejido adiposo NXY 058 Molecula hidrofílica altamente cargada

25
Dosis de mantenimiento
Consideraciones: t1/2 del medicamento Indice terapéutico Carácterísticas del paciente Efecto deseado Factores logísticos Ke, cinética de eliminación Paciente crítico tiene cambios en el Cl y en el VD MD = CL x Cp Infusión contínua vs Bolos Continuous infusion is appropriate for drugs with short half-lives, such as nitrovasodilators, esmolol, heparin, propofol, and cis-atracurium, to take advantage of the properties of titration of the drug to the desired effect and also to ease nursing responsibilities that are associated with frequent doses. Continuous infusion also may be appropriate to achieve the desired drug effect by enhancing the drug effect while minimizing the peak and trough effects that are observed with intermittent dosing. Fluctuations in plasma level may be deleterious if the therapeutic index is narrow or if physiologic rebound mechanisms are induced by subtherapeutic levels. Effects of loop diuretics and histamine (H2)-receptor antagonists are enhanced when administered as an infusion, because there is increased cumulative exposure of drug at the receptor site, as well as the minimization of physiologic rebound mechanisms of salt retention and acid production, respectively. Intermittent bolus regimens are appropriate when the half-life is long and frequency of dosing is reasonable. Intermittent dosing is also appropriate to allow for drug-free intervals. Drugs such as nitroglycerin, dobutamine, and opiates can elicit tolerance during uninterrupted administration, and may require escalating doses to achieve a constant physiologic effect. Recent evidence concerning sedation of intubated patients indicates that sedation-free intervals allow for adequate monitoring of neurologic status and decreases the time required to liberate the patient from the ventilator, probably by preventing the undetected accumulation of sedative drugs, or other clinically inapparent causes of CNS depression [3]. The preceding description of drug disposition applies to drugs that exhibit linear/first-order pharmacokinetics, as applies to most agents. Zero-order pharmacokinetics are required to describe the elimination of drugs by a saturable or capacity-limited pathway, in which case a constant amount of drug, rather than a constant fraction, is eliminated per unit time. The ‘‘half-life’’ of elimination of these agents is concentration- dependent, because of capacity-limited clearance. Small dose escalations may result in disproportionately large plasma concentration increments when the maximum metabolic capacity is exceeded. Common drugs, such as ethanol, salicylates, and phenytoin, observe zero-order kinetics, although many drugs may develop saturation of metabolic pathways in overdose.
-receptor antagonists are enhanced. when administered as an infusion, because there is. increased cumulative exposure of drug at the receptor. site, as well as the minimization of physiologic rebound. mechanisms of salt retention and acid production, respectively. Intermittent bolus regimens are. appropriate when the half-life is long and frequency. of dosing is reasonable. Intermittent dosing is also. appropriate to allow for drug-free intervals. Drugs. such as nitroglycerin, dobutamine, and opiates can. elicit tolerance during uninterrupted administration, and may require escalating doses to achieve a constant. physiologic effect. Recent evidence concerning sedation. of intubated patients indicates that sedation-free. intervals allow for adequate monitoring of neurologic. status and decreases the time required to liberate the. patient from the ventilator, probably by preventing the. undetected accumulation of sedative drugs, or other. clinically inapparent causes of CNS depression [3]. The preceding description of drug disposition. applies to drugs that exhibit linear/first-order pharmacokinetics, as applies to most agents. Zero-order pharmacokinetics. are required to describe the elimination. of drugs by a saturable or capacity-limited pathway, in. which case a constant amount of drug, rather than a. constant fraction, is eliminated per unit time. The. ‘‘half-life’’ of elimination of these agents is concentration- dependent, because of capacity-limited clearance. Small dose escalations may result in disproportionately. large plasma concentration increments when the. maximum metabolic capacity is exceeded. Common. drugs, such as ethanol, salicylates, and phenytoin, observe. zero-order kinetics, although many drugs may. develop saturation of metabolic pathways in overdose.")
26
Dosis-intervalo de administración para ampicilina
6.6 mg/kg cada 12 horas 11 mg/kg cada 6 horas Siempre considerar la farmacodinámia 15 mg/kg cada 8horas Relationship between different antimicrobial doses and dosage intervals ( τ ) against a pathogen with a high or low minimum inhibitory concentration (MIC). Dosage conditions are (A) 6.6 mg/kg every 12 h, (B) 11 mg/kg every 6 h, (C) 15 mg/kg every 8 h, and (D) 22 mg/kg every 12 h. Hatched areas of the profi les are time greater than MIC. 22mg/kg cada 12 horas
 against a pathogen. with a high or low minimum inhibitory concentration (MIC). Dosage conditions are (A) 6.6 mg/kg. every 12 h, (B) 11 mg/kg every 6 h, (C) 15 mg/kg every 8 h, and (D) 22 mg/kg every 12 h. Hatched areas. of the profi les are time greater than MIC. 22mg/kg cada 12 horas.")
27
Unión a proteinas plasmáticas
Distribución Unión a proteinas plasmáticas La fracción no unida a proteinas tiene cambios significativos en el paciente criticamente enfermo La fracción no unida se distribuye libremente a varios tejidos tisulares aumentando el VD Otras consideraciones en metabolísmo Por este tipo de alteraciones es necesario monitorizar niveles en el paciente criticamente enfermo Changes in distribution of highly protein-bound drugs are to be expected in the critically ill patient. As is discussed in more detail in the metabolism section of this article, synthesis of such proteins as a1-acid glycoprotein (AAG) and albumin undergoes significant changes. This results in altered plasma concentrations of these proteins and a corresponding change in the pharmacokinetics of highly protein-bound drugs. The general principle requiring consideration is the fraction of drug that remains unbound. As the concentration of plasma protein decreases, the concentration of protein-bound drug decreases, resulting in an increased unbound fraction. Unbound drug is free to distribute to various tissues in the body, thus increasing the volume of distribution. The reverse is true when the plasma protein concentration increases. The drugs that need to be considered based on protein binding are discussed in the metabolism section of this article.
 and. albumin undergoes significant changes. This results in altered plasma concentrations. of these proteins and a corresponding change in the pharmacokinetics of. highly protein-bound drugs. The general principle requiring consideration is the. fraction of drug that remains unbound. As the concentration of plasma protein. decreases, the concentration of protein-bound drug decreases, resulting in an. increased unbound fraction. Unbound drug is free to distribute to various tissues. in the body, thus increasing the volume of distribution. The reverse is true when. the plasma protein concentration increases. The drugs that need to be considered. based on protein binding are discussed in the metabolism section of this article.")
28
Distribución Efecto de la temperatura *Estudio en perros
Ratio > 1 preferencia en distribución Ratio < 1 distribución restringida Ratio 1 No preferencia

29
Flujo sanguíneo hepático
Metabolísmo Flujo sanguíneo hepático Actividad Enzimática Unión a proteinas CLH=Q x E E = fu x CLint/[Q + fu x CLint] Razón de extracción: Alto: >0.7 Intermedio: 0.3 – 0.7 Bajo: <0.3 Hepatic metabolism depends primarily on three physiologic processes: hepatic blood flow (HBF), enzyme activity, and protein binding. Alterations in one or more of these processes result in varying effects on hepatic metabolism depending on the characteristics of the drug. The general equation describing the hepatic clearance of drugs is CLH=Q d E, where CLH, Q, and E represent total hepatic drug clearance, total HBF, and the hepatic extraction ratio, respectively. The extraction ratio, in turn, is dependent on the drug-metabolizing capabilities of the hepatic enzymes and the protein-binding characteristics of the drug. Specifically, the extraction ratio can be expressed as E = fu d CLint/[Q + fu d CLint], where fu is the unbound fraction of drug and CLint is the intrinsic hepatic clearance or the maximum metabolizing capability of the liver [40]. Extraction ratios can be generally classified as high (N0.7), intermediate (0.3–0.7), and low (b0.3) according to the fraction of drug removed during one pass through the liver. Knowledge of the hepatic extraction ratio for a particular drug is useful in predicting changes in drug metabolism because it relates to changes in HBF, enzyme activity, and protein binding.
, enzyme activity, and protein binding. Alterations in one or. more of these processes result in varying effects on hepatic metabolism. depending on the characteristics of the drug. The general equation describing. the hepatic clearance of drugs is CLH=Q d E, where CLH, Q, and E represent. total hepatic drug clearance, total HBF, and the hepatic extraction ratio, respectively. The extraction ratio, in turn, is dependent on the drug-metabolizing. capabilities of the hepatic enzymes and the protein-binding characteristics of the. drug. Specifically, the extraction ratio can be expressed as E = fu d CLint/[Q + fu d. CLint], where fu is the unbound fraction of drug and CLint is the intrinsic hepatic. clearance or the maximum metabolizing capability of the liver [40]. Extraction. ratios can be generally classified as high (N0.7), intermediate (0.3–0.7), and low. (b0.3) according to the fraction of drug removed during one pass through the. liver. Knowledge of the hepatic extraction ratio for a particular drug is useful in. predicting changes in drug metabolism because it relates to changes in HBF, enzyme activity, and protein binding.")
30
Relación fracción de extracción y flujo hepático

31
Ejemplos de medicamentos
7.6.1 Low e xtraction When metabolic capacity is low (low Cl int ), the drug is defi ned as being a low - extraction drug, and its clearance will be dependent on the extent of protein binding. In the example of protein binding displacement in Chapter 5 , a sixfold increase in the free fraction of drug could increase hepatic metabolism by a similar amount, which paradoxically would eliminate any potential adverse effect. A low - extraction drug ’ s clearance will be independent of hepatic blood fl ow. Low - extraction drugs generally have inadequate quantities of enzyme, poor biliary transport, or poor diffusion of the drug to the site of metabolism. The disposition of these drugs is also susceptible to enzyme induction (which would increase Cl int ) and further enzyme inhibition. 7.6.2 High e xtraction In contrast, if Cl int is high, the drug is considered to be a high - extraction compound, and its clearance is now limited to how much drug can be delivered to the organ, hence its hepatic blood fl ow. Changes in protein binding will not affect its clearance . High - extraction drugs will also have a high fi rst - pass effect after oral administration and, in many cases, may prevent oral administration from being an effective route of dosing even if the compound is well absorbed across the gastrointestinal mucosa. For these drugs, enzyme induction will have little effect; however, enzyme inhibition may decrease Cl int suffi ciently to decrease the extraction ratio to the range of a moderate - to low - extraction drug. Table 7.12 lists drugs according to extraction ratios and, for comparison, also lists drugs in a similar classifi cation that are cleared by the kidney. These factors are important to consider when making interspecies extrapolations (Chapter 18 ) and when adjusting dosage regimens for renal or hepatic disease (Chapter 17 ).
, the drug is defi ned as being a low - extraction. drug, and its clearance will be dependent on the extent of protein binding. In the example. of protein binding displacement in Chapter 5 , a sixfold increase in the free fraction of drug. could increase hepatic metabolism by a similar amount, which paradoxically would eliminate. any potential adverse effect. A low - extraction drug ’ s clearance will be independent of. hepatic blood fl ow. Low - extraction drugs generally have inadequate quantities of enzyme, poor biliary transport, or poor diffusion of the drug to the site of metabolism. The disposition. of these drugs is also susceptible to enzyme induction (which would increase Cl int ) and further enzyme inhibition High e xtraction. In contrast, if Cl int is high, the drug is considered to be a high - extraction compound, and. its clearance is now limited to how much drug can be delivered to the organ, hence its. hepatic blood fl ow. Changes in protein binding will not affect its clearance . High - extraction. drugs will also have a high fi rst - pass effect after oral administration and, in many cases, may prevent oral administration from being an effective route of dosing even if the compound. is well absorbed across the gastrointestinal mucosa. For these drugs, enzyme induction. will have little effect; however, enzyme inhibition may decrease Cl int suffi ciently to. decrease the extraction ratio to the range of a moderate - to low - extraction drug. Table 7.12 lists drugs according to extraction ratios and, for comparison, also lists drugs. in a similar classifi cation that are cleared by the kidney. These factors are important to. consider when making interspecies extrapolations (Chapter 18 ) and when adjusting dosage. regimens for renal or hepatic disease (Chapter 17 ).")
32
Flujo sanguíneo hepático
Metabolísmo Flujo sanguíneo hepático Variaciones aumentan o disminuyen la Cl hepática Se afetan más los que tengan alta fracción de extracción: Ejemplo midazolam, bbloqueadores, lidocaina.. Los de alta extracción son flujo dependientes Sepsis: estado hiperdinámico vs hipodinámico Estados de falla: Shock, IAM, falla cardiaca bajan Cl hepático Ventilación mecánica, vasoconstrictores, vasodilatadores Alterations in HBF can affect drug metabolism by increasing or decreasing drug delivery to the hepatocyte. The most clinically important group of drugs would be those that are highly extracted by the liver (E N0.7). In other words, hepatic metabolism of high hepatic extraction ratio drugs is dependent on HBF and relatively unaltered by changes in hepatic enzyme activity. This occurs because the drug has sufficient time to dissociate from blood components, enter the hepatocyte, and undergo biotransformation or biliary excretion. The efficiency of this process is so great that hepatic perfusion becomes the rate- 260 boucher et al limiting process in the hepatic metabolism of high extraction. Examples of intermediate- and high-extraction drugs used in the critically ill patient include lidocaine, beta-blockers, morphine, and midazolam. Sepsis is commonly manifested in critically ill patients and can lead to profound changes in HBF for high-extraction drugs. During the hyperdynamic stage of sepsis, cardiac output (CO) typically increases and blood flow distribution changes to shunt blood flow to vital organs. The opposite is true during late sepsis, where HBF reductions may decrease the clearance of these compounds. Hemorrhagic and other forms of hypovolemic shock, myocardial infarction, and acute heart failure are other problems in critically ill patients in which one can anticipate a decrease in drug clearance for high-extraction drugs. Numerous animal and clinical studies have investigated this phenomenon and have generally confirmed the expected effects of these conditions on HBF, as summarized in a comprehensive review of this topic by McKindley and colleagues [41]. In addition to the effect of critical illness on HBF, iatrogenically induced alterations in HBF may lead to changes in the elimination of intermediate- to high-extraction compounds. Such conditions include the use of mechanical ventilation with or without the administration of positive end-expiratory pressure (PEEP), which is often required in critically ill patients to facilitate delivery of oxygen and gas exchange [42]. Furthermore, drugs may also affect HBF, which could produce significant alterations in the clearance of other drugs whose elimination has blood flow–dependent characteristics. In general, a-adrenoceptor agonists, such as phenylephrine, norepinephrine, epinephrine, and dopamine (N10–12 mg/kg/min), can produce hepatic arterial and portal vein vasoconstriction, leading to decreased total HBF [43]. Vasopressin also has the potential for deceasing HBF [44]. Conversely, nitroglycerin may increase HBF by decreasing portal and hepatic vein resistance. Inotropes like dopamine and dobutamine have been shown to increase HBF by increasing CO. Antihypertensive agents seem to have variable effects on HBF.
. In other words, hepatic metabolism of high hepatic extraction ratio drugs is dependent on HBF. and relatively unaltered by changes in hepatic enzyme activity. This occurs. because the drug has sufficient time to dissociate from blood components, enter. the hepatocyte, and undergo biotransformation or biliary excretion. The. efficiency of this process is so great that hepatic perfusion becomes the rate- 260 boucher et al. limiting process in the hepatic metabolism of high extraction. Examples of. intermediate- and high-extraction drugs used in the critically ill patient include. lidocaine, beta-blockers, morphine, and midazolam. Sepsis is commonly manifested in critically ill patients and can lead to profound. changes in HBF for high-extraction drugs. During the hyperdynamic stage. of sepsis, cardiac output (CO) typically increases and blood flow distribution. changes to shunt blood flow to vital organs. The opposite is true during late sepsis, where HBF reductions may decrease the clearance of these compounds. Hemorrhagic and other forms of hypovolemic shock, myocardial infarction, and. acute heart failure are other problems in critically ill patients in which one can. anticipate a decrease in drug clearance for high-extraction drugs. Numerous. animal and clinical studies have investigated this phenomenon and have generally. confirmed the expected effects of these conditions on HBF, as summarized in a. comprehensive review of this topic by McKindley and colleagues [41]. In addition to the effect of critical illness on HBF, iatrogenically induced. alterations in HBF may lead to changes in the elimination of intermediate- to. high-extraction compounds. Such conditions include the use of mechanical. ventilation with or without the administration of positive end-expiratory pressure. (PEEP), which is often required in critically ill patients to facilitate delivery of. oxygen and gas exchange [42]. Furthermore, drugs may also affect HBF, which. could produce significant alterations in the clearance of other drugs whose. elimination has blood flow–dependent characteristics. In general, a-adrenoceptor. agonists, such as phenylephrine, norepinephrine, epinephrine, and dopamine. (N10–12 mg/kg/min), can produce hepatic arterial and portal vein vasoconstriction, leading to decreased total HBF [43]. Vasopressin also has the potential for. deceasing HBF [44]. Conversely, nitroglycerin may increase HBF by decreasing. portal and hepatic vein resistance. Inotropes like dopamine and dobutamine have. been shown to increase HBF by increasing CO. Antihypertensive agents seem to. have variable effects on HBF.")
33
Depuración intrínsica hepática
Metabolísmo Depuración intrínsica hepática Metabolísmo de fase I más importancia clínica que fase II Pacientes con SIRS: las catecolaminas inhiben CYPs Pacientes de TEC: Aumento de metabolísmo y disminución de efecto de anticonvulsivantes Se recomienda suplemento proteico agresivo Importantes excepciones morfina y clindamicina que requieren glucoronidacion McKindley DS, et al. Hepatic drug metabolism in critical illness. Pharmacotherapy 1998;18(4):759–78.
:759–78.")
34
Interacciones en CYPs

35
Hinderling, T. et al. Ther Drug Monit 2005;27:71–85

36
Revisión de mediciones de influencia del pH en unión a proteinas
Hinderling, P. et al. Ther Drug Monit 2005;27:71–85

37
The 25 drugs in a list of 456 drugs14,15 for which protein binding may influence clinical drug exposure after nonoral administration, with use of cutoffs of >70% for protein binding (fu < 0.3) and 0.28 Qorgan for clearance
 and 0.28 Qorgan for clearance")
38
Unión a proteínas Metabolísmo Stress disminuye Albúmina y aumenta AAG
En quemados aumento de 2 a 3veces AAG y disminución de 2 veces la Albúmina Medicamentos acídicos (diazepem) que se unen a la albúmina aumentan fracción libre Medicamentos básicos que se únen más a AAG (meperidina, propranolol, lidocaina) disminuye su fracción libre Alterations in protein binding primarily affect the hepatic clearance of lowextraction drugs, because high-extraction drugs are completely metabolized independent of protein binding (nonrestrictive hepatic metabolism). In general, hepatic metabolism of low-extraction drugs is restrictive, meaning that metabolism is limited to the unbound fraction. Because only unbound drug is able to diffuse into the hepatocyte, for low-extraction drugs, the fraction unbound correlates with the rate of elimination. The overall importance of alterations in protein binding in the critically ill patient involves the proper interpretation of measured drug concentrations and their pharmacodynamic effect, because only unbound drug is free to interact with its corresponding receptor. Thus, knowledge of the extraction ratio is essential to predicting the pharmacokinetic outcome resulting from protein-binding changes. It has been demonstrated in critically ill patients that albumin concentrations decrease and AAG synthesis increases during and after traumatic or physiologic stress. This has been demonstrated in multiple critically ill patient subsets. As a result, the pharmacokinetics of albumin-bound or AAG-bound drugs may change. For example, patients with thermal injury demonstrated a two- to threefold increase in AAG concentrations and a twofold decrease in albumin concentrations that lasted the entire 1-month study period [55]. As a result, the fraction unbound increased for acidic drugs primarily bound to albumin (eg, phenytoin, diazepam) but decreased for basic drugs primarily bound to AAG (eg, meperidine, propranolol, lidocaine). This emphasizes the need to monitor the free or unbound concentrations of highly bound drugs in the critically patient. Conversely, the pharmacologic response to drugs highly bound to AAG can be changed dramatically. The unbound fraction of lidocaine decreased from 28% to 15% as AAG concentrations increased in one clinical study. As a result, higher total concentrations of lidocaine were required to achieve pharmacologic effects and were tolerated without toxic effects, because more lidocaine was protein bound and unable to exert pharmacologic effects [56]. Although the overall number of agents for which protein-binding alterations significantly affect drug exposure has been found to be limited based on a recent systematic review, several agents are routinely administered to critically ill patients [57]. In addition to those already addressed, this list includes fentanyl, alfentanil, sufentanil, remifentanil, diltiazem, nicardipine, verapamil, erythromycin, haloperidol, itraconazole, milrinone, and propofol [57].
 que se unen a la albúmina aumentan fracción libre. Medicamentos básicos que se únen más a AAG (meperidina, propranolol, lidocaina) disminuye su fracción libre. Alterations in protein binding primarily affect the hepatic clearance of lowextraction. drugs, because high-extraction drugs are completely metabolized. independent of protein binding (nonrestrictive hepatic metabolism). In general, hepatic metabolism of low-extraction drugs is restrictive, meaning that. metabolism is limited to the unbound fraction. Because only unbound drug is. able to diffuse into the hepatocyte, for low-extraction drugs, the fraction unbound. correlates with the rate of elimination. The overall importance of alterations in. protein binding in the critically ill patient involves the proper interpretation of. measured drug concentrations and their pharmacodynamic effect, because only. unbound drug is free to interact with its corresponding receptor. Thus, knowledge. of the extraction ratio is essential to predicting the pharmacokinetic outcome. resulting from protein-binding changes. It has been demonstrated in critically ill patients that albumin concentrations. decrease and AAG synthesis increases during and after traumatic or physiologic. stress. This has been demonstrated in multiple critically ill patient subsets. As a. result, the pharmacokinetics of albumin-bound or AAG-bound drugs may. change. For example, patients with thermal injury demonstrated a two- to. threefold increase in AAG concentrations and a twofold decrease in albumin. concentrations that lasted the entire 1-month study period [55]. As a result, the. fraction unbound increased for acidic drugs primarily bound to albumin. (eg, phenytoin, diazepam) but decreased for basic drugs primarily bound to AAG. (eg, meperidine, propranolol, lidocaine). This emphasizes the need to monitor the. free or unbound concentrations of highly bound drugs in the critically patient. Conversely, the pharmacologic response to drugs highly bound to AAG can be. changed dramatically. The unbound fraction of lidocaine decreased from 28% to. 15% as AAG concentrations increased in one clinical study. As a result, higher. total concentrations of lidocaine were required to achieve pharmacologic effects. and were tolerated without toxic effects, because more lidocaine was protein. bound and unable to exert pharmacologic effects [56]. Although the overall. number of agents for which protein-binding alterations significantly affect drug. exposure has been found to be limited based on a recent systematic review, several agents are routinely administered to critically ill patients [57]. In addition. to those already addressed, this list includes fentanyl, alfentanil, sufentanil, remifentanil, diltiazem, nicardipine, verapamil, erythromycin, haloperidol, itraconazole, milrinone, and propofol [57].")
39
Recomendaciones en enfermedad hepática

40
Excreción Depuración de medicamentos o metabolitos dependiente de función renal: Compromiso multifactorial en pacientes críticos Pacientes críticos con falla renal vs pacientes sin falla renal In addition, some drugs have active or partially active metabolites that are renally cleared and thus can accumulate in renal dysfunction. Renal dysfunction in critically ill patients can present as preexisting chronic renal failure, new-onset acute renal failure commonly attributable to hypoperfusion or tubular necrosis, or a combination of both. Dosing recommendations for patients with varying degrees of renal dysfunction are widely available from manufacturers’ prescribing information, tertiary drug references, and the primary literature. The need for dialysis, the type of dialysis (intermittent versus continuous), and the frequency of dialysis should also be considered. Dosing recommendations for patients requiring dialysis are also available from these sources, albeit with fewer data for newer continuous renal replacement therapies [58]. Thus, the focus of this section is on alterations in renal drug clearance or tO in critically ill patients with apparently normal renal function.
, and the frequency of dialysis should also be considered. Dosing. recommendations for patients requiring dialysis are also available from these. sources, albeit with fewer data for newer continuous renal replacement therapies. [58]. Thus, the focus of this section is on alterations in renal drug clearance or tO. in critically ill patients with apparently normal renal function.")
41
Pacientes críticos sin falla renal:
Excreción Pacientes críticos sin falla renal: Potencial de niveles subterapeuticos vs tóxicos

42
Relación vida media vs Cl-Creatinina

43
Efecto de la terapia de remplazo renal
Cuando ajustar dosis Cuando dar dosis extra Medicamentos dializables

44
Excreción Pacientes con TRR

45
Monitoreo de niveles terapéuticos
Alta variabilidad Niveles pico y valle Anticonvulsivantes, inmunosupresores, antibioticos, digoxina Metabolismo tipo Hoffman preferidos

46
Factores que disminuyen
Conclusiones Factores que aumentan Factores que disminuyen Disfunción renal Disfunción hepática Disfunción cardiaca GC aumentado Aumento en el Vd Absorción disminuida Aproximación multidisciplinaria, valorar riesgo beneficio Algunos medicamentos requieren corrección de dosis Algunas alteraciones pueden no tener impacto clínico..

47
Gracias.

48
Power, B. et al. Pharmacokinetics of Drugs Used in Critically Ill Adults, Clin Pharmacokinet 1998 Jan; 34 (1): 25-56
:")
Presentaciones similares