Descargar la presentación
La descarga está en progreso. Por favor, espere

1
CASO CLINICO NEUROPEDIATRIA
JANNETH MARCELA TEHERAN Residente de Pediatría DR. MARIA EUGENIA MIÑO Mayo de 2009

2
HISTORIA CLINICA Paciente de 8 años Procedente de Popayán.
Fecha ingreso: 20- abril horas Fuente información: madre

3
HISTORIA CLINICA Un día antes al ingreso el paciente es arroyado por una bicicleta en horas de la noche, presenta pérdida del conocimiento por 5 minutos, la madre consulta a hospital de II nivel en donde realizan manejo con medidas generales curación de heridas y lo dejan en observación por 8 horas. Buena evolución se da salida a las 3 am. con recomendaciones y control médico, según la madre el paciente estaba dormido al salir del hospital.

4
HISTORIA CLINICA La madre refiere que al día siguiente que el niño permanece somnoliento durante todo el día, con imposibilidad para caminar, cefalea que calma con analgesia, alteración en la marcha y perdida del equilibrio por lo cual consulta.

5
HISTORIA CLINICA Antecedentes personales: madre de G2P2, CPN # 4, embarazo sin hospitalizaciones, nació a término, parto domiciliario “no complicado”. Desarrollo psicomotor: sostén cefálico 2 años, gateó 1 año, caminó a los 2 ½ años, tiene problemas en la marcha, se cae con frecuencia el padre refiere que en este año ha mejorado. Escolar en 1 grado, ha perdido 3 veces el primer año, tiene dificultades en el aprendizaje, pero no es indisciplinado en el colegio, no sabe leer ni escribir.

6
HISTORIA CLINICA Antecedentes patológicos:
En 3 años le encontraron “cabeza grande” se estudio por consulta externa de pediatría y le tomaron un TAC cerebral, al parecer es normal pero el padre no volvió a control.

7
EXAMEN FISICO FC: 62 FR:20 T:36.5° TA: 120/80 SO2:96%
P: 25kg (p15-50) T: 119cm(p3-15) PC:58cm. P>90% Neurológico: alerta, orientado, Glasgow 15/15, lenguaje claro, pupilas isocóricas fotorreactivas pares craneanos normales, tono normal, marcha inestable, hiperreflexia en miembros inferiores Asimetría en cráneo con dolor a la palpación en lado derecho de la cara, escoriaciones, orejas bien implantadas.
 T: 119cm(p3-15) PC:58cm. P>90% Neurológico: alerta, orientado, Glasgow 15/15, lenguaje claro, pupilas isocóricas fotorreactivas. pares craneanos normales, tono normal, marcha inestable, hiperreflexia en miembros inferiores. Asimetría en cráneo con dolor a la palpación en lado derecho de la cara, escoriaciones, orejas bien implantadas.")
8
EXAMEN FÍSICO Corazón rítmico sin soplos, pulsos simétricos, presión normal. Murmullo vesicular simétrico, no roncus ni estertores. Abdomen no distendido, blando no doloroso, no masas. Extremidades sin edemas.

9
HISTORIA CLINICA Trauma craneoencefálico moderado
DIAGNÓSTICO Trauma craneoencefálico moderado Trauma facial en tejidos blandos Retraso en desarrollo psicomotor Escolar con estado nutricional normal Macrocefalea de etiología a estudio. Se deja en observación con líquidos TAC cerebral simple: importante atrofia cortical

10
HISTORIA CLINICA NEUROPEDIATRIA.
Tomografía muestra sustancia blanca con hipodensidad, mayor en región frontal lo que sugiere compromiso de sustancia blanca compatible con LEUCODISTROFIA METACROMATICA Se solicita cuantificación de arilfosfatasa y B- galactosidasa, RMN.

11
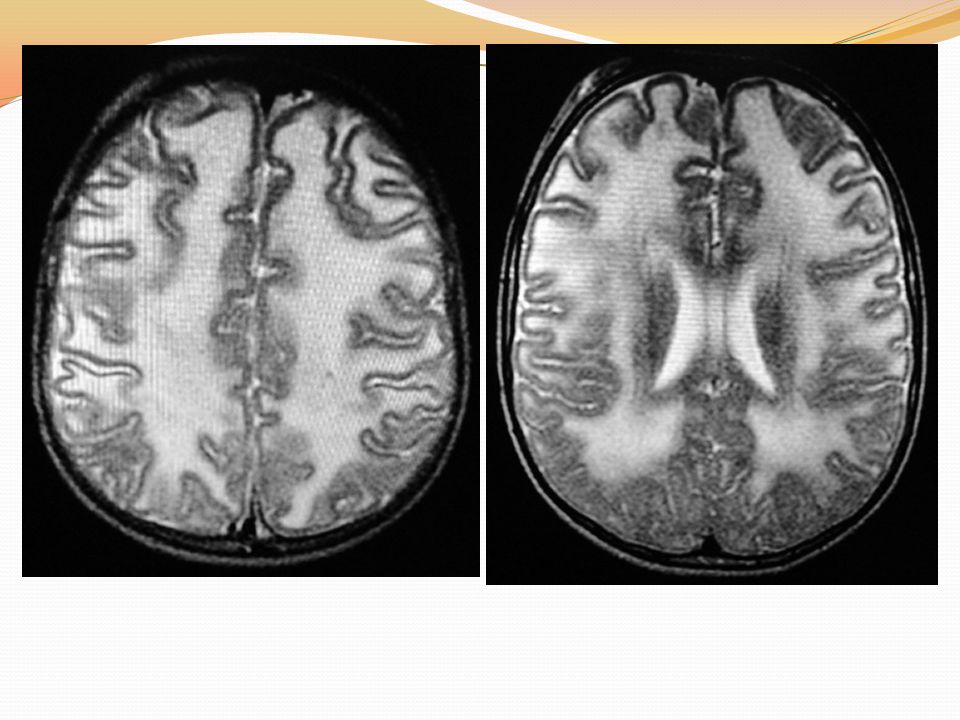
Resonancia magnética nuclear

12
Resonancia magnética nuclear

13
Resonancia magnética nuclear

14
Resonancia magnética nuclear

16
HISTORIA CLINICA RMN: se observa compromiso de la sustancia blanca predominante en regiones frontales y occipitales Se da salida, valoración por fisiatría Control en 1 mes.

17
CASO CLINICO 2

18
CASO CLINICO 2 Edad: 11 años. Procedente de Corinto Fecha: 31 de enero. 19 horas. Remitida de primer nivel con diagnóstico estatus convulsivo.

19
Historia clínica Paciente con historia de convulsiones a los 2 meses de edad, fue hospitalizada pero no recibió tratamiento, ni control. Desde hace 2 años convulsiones tonicoclónicas , manejadas con ácido valpróico desde entonces presenta crisis cada dos meses, desde hace 1 años la madre nota marcha parética y desde 2 meses las convulsiones se presentan diariamente, se evidencia hemiparesia izquierda progresiva, acompañada con cefalea intensa, emesis. El día del ingreso presenta 4 episodios, consultan a I nivel, aplican Valium, fenitoina y remiten.

20
Antecedentes personales
Madre de 40 años, G5 P4 A1, parto institucional, podálico, sin complicaciones, nació a término. Lactancia 1 año. Vacunación completa. Desarrollo psicomotor: normal. El año pasado estudio en 6 grado, con buen rendimiento. No antecedentes familiares de convulsiones.

21
Examen físico TA: 90/50 FC: 63 FR:28 T:36.5 PC: 53 cm Neurológico: cráneo normocéfalo, pupilas anisocóricas, derecha de 2mm, izquierda de 3mm que no responde a la luz, ptosis palpebral izquierda con desviación ocular inferior interna, parálisis facial central izquierda, hemiplejía izquierda, Babinski +, reflejo patelar ausente. Resto del examen: se describe normal.

22
Diagnóstico Síndrome convulsivo Síndrome de HTE
Proceso expansivo intracerebral. Neoplasia cerebral supratentorial. Conducta: se inician líquidos, Acido valpróico TAC cerebral y Valoración por neuropediatría

23
Diagnóstico TAC cerebral. evidencia masa expansiva peri ventricular que hace efecto de masa, no vascularización, signo de edema cerebral. se solicita TAC contrastado y valoración neuropediatría.

24
Neurocirugía 1 febrero . Paciente con convulsiones generalizadas desde hace 2 años, hace 2 meses hemiparesia izq, hace 1 semana aumento de convulsiones. Examen: alerta, glasgow 15/15, hemiparesia izquierda, pupilas isocóricas reactivas. TAC: lesión expansiva tracto temporal derecha DX: Astrocitoma de bajo grado Pendiente realizar biopsia.

25
Historia clínica 2 febrero.
Cuadro de estatus convulsivo, ya resuelto, manejado con Midazolam y Ácido Valpróico y recibe dexametasona, por sospecha de masa intracraneana. Según el tío 4 episodios convulsivos a los 2 años de edad. A los 9 años nuevamente presenta convulsiones, estudiados en Cali con EEG y TAC cerebral se inicio ácido valpróico que se suspendió hace 1 año, la paciente no conoce la causa. Desde hace 1 año presenta disminución de la fuerza en hemicuerpo derecho, episodios de movimiento tonicoclónicos generalizados con pérdida de conciencia, precedido por llanto o tristeza. Desde hace 2 meses aumento en el número de convulsiones 2 al día.

26
Historia clínica. TAC : zona hipodensa frontotemporal derecha por debajo del núcleo caudado que comprime el ventrículo lateral derecho que no capta el medio de contraste compatible con proceso tumoral tipo Astrocitoma de bajo grado. Se sugiere realizar RNM cerebral para definir la conducta a seguir, cambio de medicación a Tegretol, continuar con dexametasona.

27
Historia clínica. Reporte TAC. 31 enero
Se observa lesión intra-axial hipodensa de contorno mal definido que compromete la región ganglio basal y brazo anterior de capsula interna derechos, el cual no realza con el contraste, presenta fenómeno expansivo con colapso del cuerno frontal del ventrículo derecho sin desviar la línea media. Conclusión: masa intraaxial que sugiere neoplasia (probablemente glioma de bajo grado)
")
28
Historia clínica. Reporte de RMN 4 febrero
Múltiples eventos de tipo isquémico con compromiso predominante de ganglios basales específicamente de los derechos y a nivel cortico-subcortical de lóbulos parietales, centros semiovales y en el lóbulo temporal derecho que pueden estar relacionados con fenómenos de vasculitis a correlacionar con datos clínicos.

29
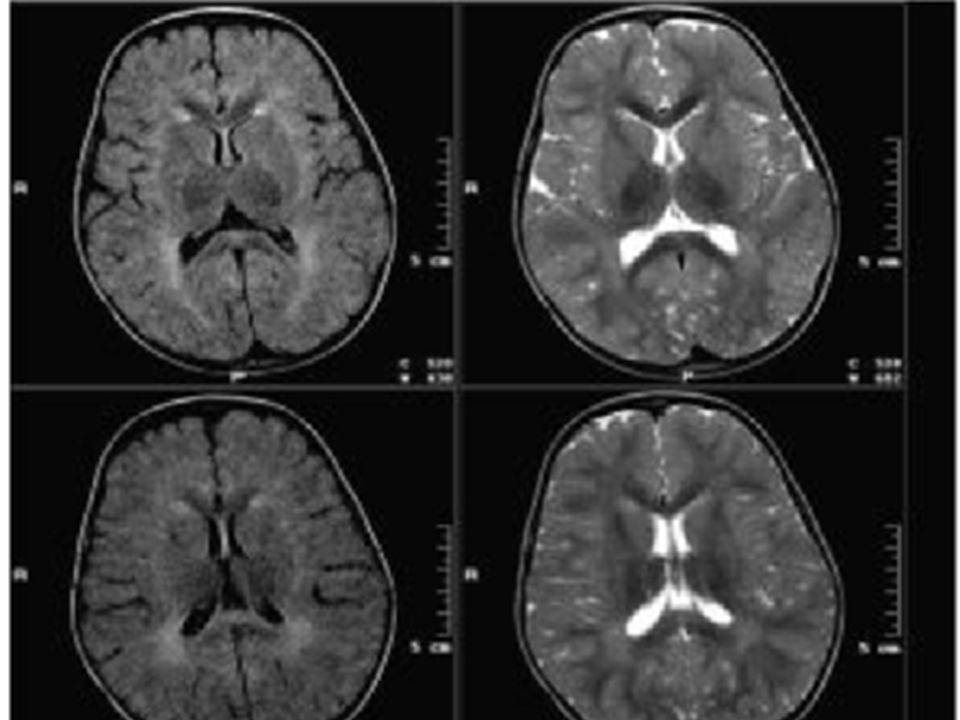
Resonancia magnética nuclear.

30
Resonancia magnética nuclear.

31
Resonancia magnética nuclear.

32
Resonancia magnética nuclear.

33
Resonancia magnética nuclear.

34
Resonancia magnética nuclear.

35
Historia clínica. 9 febrero Se revisa nuevamente RNM además de la lesión descrita, lesiones en sustancia blanca y sustancia gris temporal y parietal derecha que podría corresponder a una vasculitis vs enfermedad de sustancia blanca. 16 febrero Por curso clínico, revisión de literatura y correlación con neuroimagen, se decide iniciar manejo para esclerosis múltiple con Betaferon para aplicación SC interdiaria.

37
Leucodistrofias

38
Leucodistrofia metacromática
Frecuencia 1.4–1.8 por nv. Es la Leucodistrofia hereditaria más común. Herencia autosómica recesiva. Brazo largo cromosoma 22. Grupo O : sin actividad de la enzima Grupo R: actividad enzimática residual mínima. La homocigosis para los alelos O es > en LDM infantil tardía.

39
Leucodistrofia metacromática
La arilsulfatasa A cataliza la hidrólisis del sulfatito y se convierte en éster de sulfátido (lípido de membrana y componente de mielina) y cerebrósido. Macrófagos células de Schwann MQ en SNP Riñones, hígado, vesícula biliar, glándulas sudoríparas, páncreas, testículos, corteza adrenal y tejido rectal. Acumulación en gránulos metacromáticos que se acumulan en oligodendrocitos
 y cerebrósido. Macrófagos. células de Schwann. MQ en SNP. Riñones, hígado, vesícula biliar, glándulas sudoríparas, páncreas, testículos, corteza adrenal y tejido rectal. Acumulación en gránulos metacromáticos que se acumulan en oligodendrocitos.")
40
Leucodistrofia metacromática
FISIOPATOLOGÍA la mielina se forma normal, como la enzima esta disminuida se acumulan sulfátido y disminuyen cerebrosidos y se hace inestable y se rompe. La acumulación de sulfátidos en los lisosomas del oligodendrocito desprenda el citoplasma celular, produce tóxicos que matan la célula y el mantenimiento de la mielina no se produce. Con el curso de la enfermedad se produce gliosis reactiva y desaparecen los oligodendrocitos. La corteza esta intacta porque las neuronas no tienen los gránulos, debido a la degeneración axonal, se pierde la neurona y se produce atrofia cerebral y cerebelosa.

41
CLASIFICACIÓN 3. Forma del adulto: de 12 a los 70 años
2. Forma infantil tardía: se manifiesta entre el primer y segundo año de vida. Se presenta en 80% 1. Forma juvenil: aparece entre los 4 y los 15 años; precoz (antes de los 6 años) y tardía (después de los 6 años). 3. Forma del adulto: de 12 a los 70 años
 y tardía (después de los 6 años). 3. Forma del adulto: de 12 a los 70 años.")
42
MANIFESTACIONES CLÍNICAS
La forma infantil tardía y la juvenil precoz son los más frecuentes, tienen un fenotipo más grave y progresan rápidamente. Retraso en desarrollo psicomotor Convulsiones Arreflexia Atrofia muscular Disfagia Ceguera Sordera Cuadriparesia espástica Hipotonia seguida de parecia flacida 4 extrem.

43
MANIFESTACIONES CLÍNICAS
En la forma tardía juvenil la dificultad cognitiva, precede a los disturbios de la marcha y la progresión es más lenta, su desarrollo puede llevar más de 20ª. Dificultad en el aprendizaje escolar Trastornos en la conducta, Confusión, Dificultad en la marcha, Incontinencia, Disartria y signos extrapiramidales. La neuropatía periférica es > en F. infantiles y juveniles neuropatía desmielinizante con una grave y progresiva reducción de la velocidad de conducción en el curso de la enfermedades.

44
MANIFESTACIONES CLINICAS
En el adulto Cursa con trastornos del comportamiento y deterioro cognitivo; hay cambio de la personalidad y disminución de la capacidad intelectual, ansiedad, pensamientos desorganizados, y puede haber síntomas de depresión, psicosis, esquizofrenia o paranoia.

45
DIAGNÓSTICO Test bioquímicos
Medir la actividad ARSA para individualizar un eventual déficit. Medición de los sulfátidos en orina y el análisis molecular. La medición en cultivo de fibroblastos (varia con las características del cultivo) o por pseudodeficiencia. Es necesario, sin embargo, tener presente que una considerable variabilidad de la actividad de la enzima entre sujetos sanos y que la presencia del alelo Pd, responsable de una actividad enzimática de 5 al 15% es más bien frecuente en la población. Una actividad ARSA normal se observa después en los sujetos enfermos que poseen un déficit de saposina B, debido a que la muestra utilizada no depende de su activador, el diagnóstico de LDM podría ser erróneamente excluido
 o por pseudodeficiencia. Es necesario, sin embargo, tener presente que una considerable. variabilidad de la actividad de la enzima entre sujetos. sanos y que la presencia del alelo Pd, responsable de una actividad enzimática de 5 al 15% es más bien frecuente en la población. Una actividad ARSA normal se observa después en los sujetos enfermos que poseen un déficit de saposina B, debido a que la. muestra utilizada no depende de su activador, el. diagnóstico de LDM podría ser erróneamente excluido.")
46
DIAGNÓSTICO 1. Actividad ARSA < 10% 3. Actividad ARSA 25-60%
LDM verdadera. El análisis molecular: alelos LDM Pd y distinguir genotipos. 3. Actividad ARSA 25-60% Posible déficit del activador Saposina Orina de 24 horas para medición de sulfátidos urinarios: Bandas fuertes: déficit SAP definido. Bandas livianas: test para los alelos Pd para de diferenciar portador LDM, portador de Pd o MPd/Pd Bandas ausentes: no LDM. 2. Actividad ARSA l0-30% LDM probable. Recolección de orina de 24 horas para medición de los sulfátidos urinarios: Bandas fuertes = confirma LDM Bandas livianas = portador LDM Bandas ausentes = no LDM 4. Actividad ARSA normal Recolección de la orina de 24 horas para medición de sulfátidos urinarios: Bandas fuertes: SAP/SAP Bandas ausentes: no LDM

47
DIAGNÓTICO TAC: Disminución de coeficiente de atenuación de la sustancia blanca simétrica periventricular y en centros semiovales.

48
Diagnóstico Desmielinización simétrica y confluente de sustancia blanca periventricular y centros semiovales, con respeto de las fibras U. El comienzo es frontal y progresión fronto-occipital. Imágenes con aspecto lineal rayado, tigroide.

49
DIAGNÓSTICO Cuando la enf progresa: afecta al cuerpo calloso, cerebelo, tracto corticoespinal, capsula interna y tálamo. Patrón de desmielinización Patrón de neuropatía periférica en los pacientes con síntomas de neuropatía.

51
TRATAMIENTO Actualmente no existe ningún tratamiento disponible específico Trasplante de médula ósea en algunos casos seleccionados. Fisioterapia y fármacos antiespásticos. Fisioterapia respiratoria Adecuado aporte calórico, vitamínico y de oligoelementos.

52
PRONOSTICO La LDM es una patología degenerativa
Lleva inevitablemente a la invalidez total y a la muerte después de pocos años de aparecidos los síntomas. Pronóstico que varía entre los 5 y 10 años, en algunos casos puede extenderse a varios decenios. Complicaciones: permanencia en cama, incontinencia, incapacidad de deglutir, de comunicarse, epilepsia y a veces dolor atribuible a la neuropatía.

53
Enfermedad de Canavan Enfermedad degenerativa, autosómica recesiva
Comienza en primero días de vida 20%, 20 – 30% en primeros 2 meses. Síntomas: pobre fijación visual, irritabilidad, succión débil. Posteriormente hipotonía marcada, nistagmos, debilidad. Macrocefalia que aparece a los 6 meses de vida y afecta el 90% de los casos .

54
Enfermedad de Canavan. Manifestaciones: Hipotonía progresiva, con espasticidad, postura decorticación, retraso intelectual, atrofia óptica y muerte. Los potenciales visuales evocados y son normales, ceguera. TAC: disminución en atenuación de sustancia blanca. Se presenta por deficiencia de mielina y vacuolización generalizada de la sustancia blanca.

55
Enfermedad de Canavan Diagnóstico:
Incremento de ácido N acetil aspartico en orina, se pueden medir desde 6 semanas de edad. Disminución de actividad N-acetilaspartocilasa en cultivo de fibroblastos. Se desconoce la función

56
Enfermedad de Alexander
En casos infantiles, la enfermedad comienza en los primero 2 años Macrocefalia (algunos hidrocefalia) Regresión psicomotora Convulsiones Espasticidad Generalmente conduce a la muerte en la I década Casos juveniles: signos bulbares, ataxia y espasticidad, las habilidades intelectuales esta conservadas. Se asocia con perdida de mielina en forma rostro-caudal.
 Regresión psicomotora. Convulsiones. Espasticidad. Generalmente conduce a la muerte en la I década. Casos juveniles: signos bulbares, ataxia y espasticidad, las habilidades intelectuales esta conservadas. Se asocia con perdida de mielina en forma rostro-caudal.")
57
Enfermedad de Alexander
Casi siempre es esporádica, en pocos casos descritos en niños, se ha postulado transmisión autosómica recesiva, con hermanos severamente afectados. RMN: cambios en materia blanca con predominio frontal, anormalidades en ganglios basales, en algunos casos realce del contraste asociado con dilatación variable de ventrículos.

58
Síndrome de Leucodistrofia
Constituye el 40% de los pacientes con leucodistrofia de causa desconocida. Ataxia: síntoma inicial entre 1 – 5 años. Pueden permanecen estables por muchos años. Algunos desarrollan síntomas después de los 5 años o en adultos y sobreviven por largo tiempo: diplejía espástica lentamente progresiva, relativa de conservación de capacidad cognitiva

59
Síndrome de Leucodistrofia
Incremento en la dificultad para caminar Temblor Espasticidad Hipereflexia Disartria Convulsiones La mayoría tienen un desarrollo inicial normal y perímetro cefálico no alterado. Síntomas tardíos: Dificultades en la deglución y atrofia óptica (potenciales auditivos E. son normales). deterioro progresivo
. deterioro progresivo.")
60
Síndrome de Leucodistrofia
El electrodiagnóstico del SNP es normal. TAC: hipodensidad hemisférica difusa y simétrica en la materia blanca. Ausencia de atrofia cortical y dilatación ventricular. RMN: disminución difusa de la señal de materia blanca en T1 e incremento en T2

62
Esclerosis múltiple

63
Esclerosis múltiple Afecta a un millón de individuos en todo el mundo. Se presenta en 1 /1000 personas. 2-4% en familiares en primer grado, monocigoticos 30%. Relación 2:1 Típicamente se presenta entre 18 – 45 años. El riesgo de desarrollar la enfermedad se relaciona con factores genético y medioambietales, la susceptibilidad probablemente es poligénica.

64
Esclerosis múltiple Se caracteriza por la tríada de inflamación, desmielinización y gliosis (tejido cicatrizal) Su evolución puede incluir recidivas-remisiones, o ser progresiva. Segundo lugar en frecuencia después de los traumatismos discapacidad neurológica en la vida adulta temprana a media.

65
Esclerosis múltiple FISIOPATOLOGÍA
La proteína básica de la mielina y glucoproteína oligodendrocitaria se comportan, como antígenos para las células T y B. Ocurre bloqueo de la conducción cuando el impulso no puede atravesar el segmento desmielinizado (la membrana del axón se hiperpolariza por la exposición de canales de potasio)
")
66
Manifestaciones clínicas
Puede comenzar en forma repentina o insidiosa. Los síntomas intensos o insignificantes Motores: Debilidad en las extremidades, espasticidad, hiperreflexia y Babinsky. Neuritis óptica, diplopía, nistagmos. Sensitivas: Parestesias, hipestesia, sensaciones desagradables, dolor. Ataxia Disfunción vesical e intestinal, estreñimiento. Disfunción cognitiva, depresión. Fatiga Síntomas paroxísticos

67
Manifestaciones clínicas

68
Clasificación

69
Diagnóstico Son lesiones que en la sustancia blanca o 1-3 lesiones periventriculares, se ubican perpendiculares a la ventrículos, son ovoides, se ve en corte sagital en T2 La secuencia FLAIR de RM es el más útil para identificar lesiones en los hemisferios cerebrales. En la médula espinal las lesiones en forma de cigarro

70
Diagnóstico

71
Tratamiento Esteroide
Interferón beta han ha demostrado ser efectiva en múltiples ámbitos clínico y radiográfico, incluida la reducción de la tasa de recaída, gravedad de recaída, la mejora de las lesiones en la RM, y prolongar el tiempo hasta la progresión a discapacidad Otros.

72
PRONOSTICO Quince años después del comienzo, sólo 20% de ellos no tendrán limitaciones funcionales; la mitad habrán avanzado hasta SPMS y necesitarán auxilio en la ambulación. Se sabe que 25 años después del comienzo, más de 80% de los pacientes de MS habrán alcanzado tal nivel de discapacidad

Presentaciones similares