Descargar la presentación
La descarga está en progreso. Por favor, espere

1
SARA BELTRÁN GARCÍA ENFERMEDADES METABÓLICAS. HUMS 18 MAYO 2011
FENILCETONURIA E HIPERFENILALANINEMIA. DEL LABORATORIO A LA CLÍNICA. EXPERIENCIA EN ARAGÓN SARA BELTRÁN GARCÍA ENFERMEDADES METABÓLICAS. HUMS 18 MAYO 2011

2
PROCESO ES NECESARIA LA EXISTENTE Y EXCELENTE COMUNICACIÓN Y RELACIÓN ENTRE EL LABORATORIO Y LOS CLÍNICOS

4
Historia 1934 A. Fölling describió por 1ª vez enfermedad en dos hermanos con: Retraso Mental + Olor especial. HAR. “Idiocia fenilpurívica” 1937 Penrose y Quastel Fenilcetonuria (PKU). Bickel restricción de Phe dieta. 1961 test Gutrie. Dx precoz, prevenir retraso mental. 1980 se describe SPKUM 1983 Woo gen PAH cromosoma 12 (q22-q24.1) 1999 Kure nuevo tratamiento BH4
. Bickel restricción de Phe dieta test Gutrie. Dx precoz, prevenir retraso mental se describe SPKUM Woo gen PAH cromosoma 12 (q22-q24.1) 1999 Kure nuevo tratamiento BH4.")
5
La incidencia global es de 1/10.000 recién nacidos.
FENILALANINA(Phe) TIROSINA (Tyr) La incidencia global es de 1/ recién nacidos. En España la incidencia estimada es de 1:19747 para PKU y de 1: la de las HPA moderadas y benignas. FENILALANINA >120 nmol/mL HIPERFENILALANINEMIA PAH 98% BH4 2%
 TIROSINA (Tyr) La incidencia global es de 1/ recién nacidos. En España la incidencia estimada es de 1:19747 para PKU y de 1: la de las HPA moderadas y benignas. FENILALANINA >120 nmol/mL HIPERFENILALANINEMIA. PAH 98% BH4 2%")
7
CLASIFICACION PKU CLÁSICA: Phe dx >1200 nmol/mL (> 20 mg/dl)
PKU MODERADA: Phe dx nmol/mL ( mg/dl) PKU SUAVE: nmol/mL (6-10 mg/dL) HFA BENIGNA: nmol/mL (2-6 mg/dL) Tolerancia de Phe (mg/dia) diferente y la actividad residual enzimática
 PKU SUAVE: nmol/mL (6-10 mg/dL) HFA BENIGNA: nmol/mL (2-6 mg/dL) Tolerancia de Phe (mg/dia) diferente y la actividad residual enzimática.")
8
PKUC 250 – 350 PKUM 350 – 400 PKUS 400 – 600 HPAB > 600
Fenotipo % actividad residual PAH Cifras Phe al diagnóstico Tolerancia Phe (mg/kg/día) Tolerancia media a Phe (mg/día) Prevalencia en España PKUC < 1% > 20 < 20 250 – 350 15.7% PKUM 1 – 5% > 10 – 20 20 – 25 350 – 400 9.0% PKUS > 5% > 6 – 10 25 – 50 400 – 600 23.0% HPAB 2.5 – 6 > 50 > 600 47.7% *Trefz FK, Bartholomé K, Bickel H, Lutz P, Schmidt H, Seyberth HW. In vivo residual activities of the phenylalanine hydroxylating system in phenylketonuria and variants. J Inherit Metab Dis 1981; 4:
 Tolerancia media a Phe. (mg/día) Prevalencia en España. PKUC. < 1% > 20. < – % PKUM. 1 – 5% > 10 – – – % PKUS. > 5% > 6 – – – % HPAB. 2.5 – 6. > 50. > % *Trefz FK, Bartholomé K, Bickel H, Lutz P, Schmidt H, Seyberth HW. In vivo residual activities of the phenylalanine hydroxylating system in phenylketonuria and variants. J Inherit Metab Dis 1981; 4:")
9
Clínica PKUC: Retraso mental y motor, microcefalia, epilepsia, eczema, hiperactividad y rasgos psicóticos (tendencias destructivas, automutilaciones impulsividad y ataques de agresividad.) Epilepsia generalizada (25%) o síndrome de West. Fenotipo característico: ojos, piel y cabellos claros, olor corporal especial y eccema (30%) Tratada precozmente buena evolución, CI dentro de la normalidad (inferiores a los grupos control) y con pequeñas dificultades en el aprendizaje, torpeza motriz, dificultades grafoperceptivas, hiperactividad y trastornos del sueño. Los niveles elevados de Phe plasmática durante los primeros 6 años de vida se correlacionan negativamente .
 Epilepsia generalizada (25%) o síndrome de West. Fenotipo característico: ojos, piel y cabellos claros, olor corporal especial y eccema (30%) Tratada precozmente buena evolución, CI dentro de la normalidad (inferiores a los grupos control) y con pequeñas dificultades en el aprendizaje, torpeza motriz, dificultades grafoperceptivas, hiperactividad y trastornos del sueño. Los niveles elevados de Phe plasmática durante los primeros 6 años de vida se correlacionan negativamente .")
11
Hiperfenilalaninemia maligna (2%)
Los defectos de cofactor o del metabolismo de la BH4, presentan manifestaciones clínicas mucho más severas y en ocasiones no responden al tratamiento . El síndrome de fenilcetonuria materna (SPKUM), es una embriopatía observada en hijos de madres afectas de HPA. CIR (40%) Cardiopatía congénita (12%) Microcefalia (73%) Retraso mental (92%) Dismorfias faciales (hipertelorismo, fisuras palpebrales cortas, epicanto, paladar ojival, micrognatia, filtrum largo, labio superior fino, pabellones auriculares grandes o poco desarrollados).
, es una embriopatía observada en hijos de madres afectas de HPA. CIR (40%) Cardiopatía congénita (12%) Microcefalia (73%) Retraso mental (92%) Dismorfias faciales (hipertelorismo, fisuras palpebrales cortas, epicanto, paladar ojival, micrognatia, filtrum largo, labio superior fino, pabellones auriculares grandes o poco desarrollados).")
12
TRATAMIENTO V. Normales 40-90 nmol/mL PRECOZ!! Antes del 10º día No existe un consenso unánime entre las sociedades científicas sobre la cifra de phe a partir de la cual se deben restringir las proteínas de la dieta. 240 μmol/L España AECOM 1998 Reino Unido Alemania E.E.U.U. Inicio tratamiento > 360 μmol/L > 420 μmol/L > 600 μmol/L 0-6 años < 360 μmol/L < 240 μmol/L 6-9 años < 480 μmol/L 9-12 años < 600 μmol/L < 900 μmol/L

13
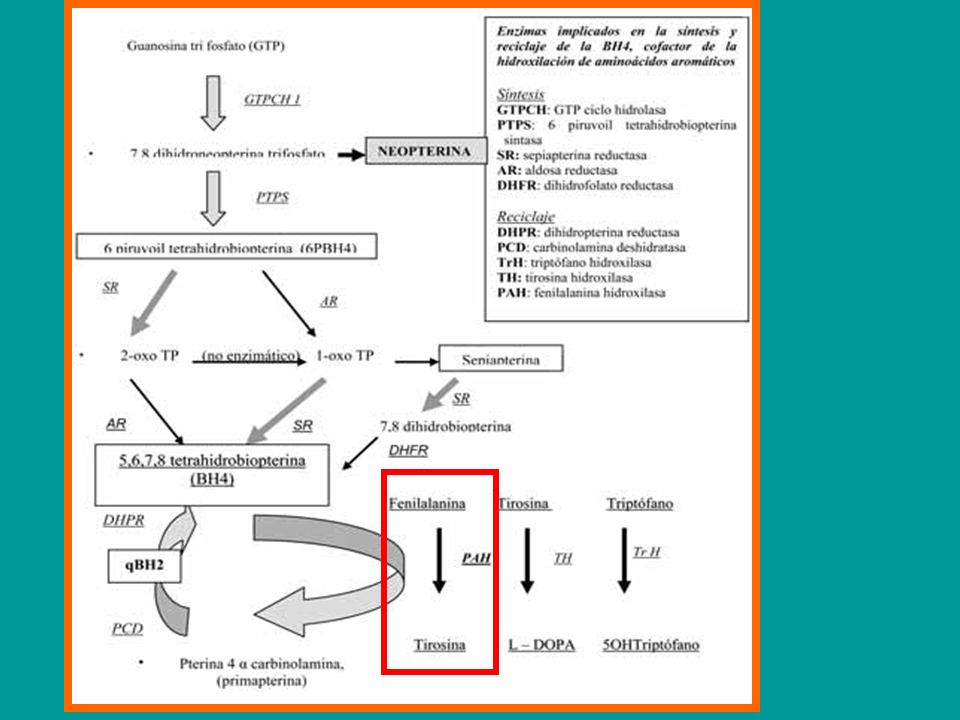
TETRAHIDROBIOPTERINA/BH4
TERAPIA GENICA TERAPIA ENZIMATICA AA LARGOS NEUTROS SOPORTE: SUPLEMENTO PROTEICO SIN PHE CALCIO LCP(W3/W6) B12... TETRAHIDROBIOPTERINA/BH4 DIETÉTICO
 B12... TETRAHIDROBIOPTERINA/BH4. DIETÉTICO.")
14
DIETETICO DIETA RESTRINGIDA EN PHE, suplementada otros aa como tyrosina, vitaminas y oligoelementos. PKU ANAMIX INFANT, PHENYL FREE 1, 2, 3 +/ L. MATERNA LACTANTES LACTANTES NIÑOS 13,1 grms prot/100gr 16,2 g de proteina/100 g g de proteina/100 g 457 kcal/100 grms Kcal/100 g kcal/100g ALIMENTOS NATURALES: Con escaso o nulo contenido proteico (<30mg/100g): frutas verduras y hortalizas. Libre administración. Contenido proteico significativo (>30 mg/100g), permitidos pero a controlar: cereales, legumbres, patatas, brócoli, espinacas, guisantes, plátanos…. Alto contenido proteico, “excluidos a priori” de la dieta, en función de la tolerancia: carnes, pescados, huevos, leche y derivados. 5% proteínas === Phe
: frutas verduras y hortalizas. Libre administración. Contenido proteico significativo (>30 mg/100g), permitidos pero a controlar: cereales, legumbres, patatas, brócoli, espinacas, guisantes, plátanos…. Alto contenido proteico, excluidos a priori de la dieta, en función de la tolerancia: carnes, pescados, huevos, leche y derivados. 5% proteínas === Phe.")
15
Alimentos MANUFACTURADOS sin Phe:
Leches harinas, pan, galletas, pasta, arroz, sucedáneo de huevo, de embutidos…. fundamentales para la confección del menú de estos niños por su variedad y calidad. APROTEN, SANAVI, LOPROFIN SUPLEMENTOS proteicos sin Phe: La dieta es hipoproteíca, es imprescindible un aporte proteíco (sin Phe). Se emplean mezclas de L-aminoácidos que en ocasiones están combinados con hidratos de carbono o grasas y enriquecidos en vitaminas y minerales.
. Se emplean mezclas de L-aminoácidos que en ocasiones están combinados con hidratos de carbono o grasas y enriquecidos en vitaminas y minerales.")
16
SHS PKU ANAMIX INFANT (13,1g prot/100 g) (XP analog LCP)
PKU ANAMIX JUNIOR LQ (10g prot/125ml=1 botella) XP MAXAMAID (25 gr prot/100 g) LOPHLEX LQ 10 (cereza, citrus naranja y tropical) (=10 g prot) LQ 20 (= 20 g prot) LOPHLEX SOBRES (1= 20g prot) ADD-INS (1 sobre=10 g prot) XP MAXAMUM (39g prot/100 g)
 XP MAXAMAID (25 gr prot/100 g) LOPHLEX LQ 10 (cereza, citrus naranja y tropical) (=10 g prot) LQ 20 (= 20 g prot) LOPHLEX SOBRES (1= 20g prot) ADD-INS (1 sobre=10 g prot) XP MAXAMUM (39g prot/100 g)")
17
ENCUESTA DIETETICA DE 3 DIAS
CALCULO: PROTEINAS TOTALES PHE (mg/kg/dia) HIDRATOS DE CARBONO GRASAS
 HIDRATOS DE CARBONO. GRASAS.")
18
FARMACOLÓGICO KUVAN ® Cofactor de la PAH
(dihidrocloruro de saproterina) Cofactor de la PAH comprimidos de 100 mg DOSIS mg/kg/día única toma al día TEST DE RESPUESTA CORTO: 48h sobrecarga phe 100mg/kg + BH4 20mg/kg control phe 0,4,8,12 y 24h Positivo <30% a las 8h o 50%a las 24h LARGO (Ensayo teraputico) 7 dias-meses
 Cofactor de la PAH. comprimidos de 100 mg. DOSIS 5-20 mg/kg/día. única toma al día. TEST DE RESPUESTA. CORTO: 48h. sobrecarga phe 100mg/kg + BH4 20mg/kg. control phe 0,4,8,12 y 24h. Positivo <30% a las 8h o 50%a las 24h. LARGO (Ensayo teraputico) 7 dias-meses.")
19
PACIENTES A TRATAR, ¿QUÉ NOS PUEDE AYUDAR?
Pacientes respondedores a la BH4 Fenotipo bioquímico (Phe al dx) Phe (dx) 900nmol/ml Genotipo (no correlacion genotipo-fenotipo) ¡OJO! NO FACTORES DETERMINANTES
 Phe (dx) 900nmol/ml. Genotipo (no correlacion genotipo-fenotipo) ¡OJO! NO FACTORES DETERMINANTES.")
20
¿CÓMO LO HACEMOS NOSOTROS?
Iniciamos sapropterina 10 mg/kg/día Misma dieta Control analítico en 1 semana 1 mes < Phe Phe > Aumentar ingesta de Phe Aumentar dosis sapropterina Control analítico en 1 semana

21
OTROS Terapia génica: Transferir mediante vectores genes de PAH (ensayo) Aminoácidos largos neutros “Bloqueo” de la absorción intestinal y del paso al sistema nervioso central de la phe, mediante el uso de suplementos orales de “aminoácidos largos neutros” (LNAA). Uso en adolescentes y adultos con fracaso en la dieta Tratamiento enzimático ● Terapia enzimática alternativa (PAL phe amonio liasa) : transforma phe en ac. transcinamico ● Trasplante hepatico: corrige el deficit de PAH, con riesgo quirurgico y de inmunosupresion
. Uso en adolescentes y adultos con fracaso en la dieta. Tratamiento enzimático. ● Terapia enzimática alternativa (PAL phe amonio liasa) : transforma phe en ac. transcinamico. ● Trasplante hepatico: corrige el deficit de PAH, con riesgo quirurgico y de inmunosupresion.")
22
RECIÉN NACIDO Ingresa para estudio
Recogida de muestras (DHPR en sangre y pterinas en orina) Inicio tratamiento dietético 24-48 horas sin proteínas Mantenemos lactancia materna Fórmulas sin Phe Valorar inicio tratamiento farmacológico (Kuvan) Controles seriados de Phe/Tyr
 Inicio tratamiento dietético horas sin proteínas. Mantenemos lactancia materna. Fórmulas sin Phe. Valorar inicio tratamiento farmacológico (Kuvan) Controles seriados de Phe/Tyr.")
23
CONTROLES PRIMER AÑO DE VIDA
CLINICO /ANALÍTICO/ DIETETICO Los 2-3 primeros meses Cada 1-2 semanas 3 meses – 6 meses Cada mes 6 meses – 1 año Cada 2-3 meses Persiste necesidad comunicación FLUIDA laboratorio/unidad de seguimiento)
")
24
CONTROLES POSTERIORES
Mes 0 Mes 3 Mes 6 Mes 9 Mes 12 P, T, PC X Desarrollo puberal Exp. Neurológica DO Desarrollo cognitivo Analítica (Phe, Tyr, AGCL) Encuesta dietéica Bioquímica hemograma hepático metabolismo hierro, perfil lipidico, aminoácidos ,Ac. Grasos, vit B12 y ac. Fólico, oligoelementos (se, zinc)
 Encuesta dietéica. Bioquímica hemograma hepático metabolismo hierro, perfil lipidico, aminoácidos ,Ac. Grasos, vit B12 y ac. Fólico, oligoelementos (se, zinc)")
25
¿?

26
¿A QUIÉN DEBE MANTENERSE EL TRATAMIENTO A LARGO PLAZO?
¿A todos en los que aumenta la tolerancia a la Phe? ¿A los que el tratamiento permite una dieta libre? ¿CUÁNTO TIEMPO DEBE MANTENERSE EL TRATAMIENTO? ¿De por vida? ¿Hasta la edad adulta? ¿A QUÉ EDAD DEBE INICIARSE EL TRATAMIENTO? < 4 AÑOS ¿Recién nacido? ¿Al año de vida? ¿En cualquier edad tras el diagnóstico?

28
DE CALIDAD DE VIDA VALIDADAS
NECESARIAS ENCUESTAS DE CALIDAD DE VIDA VALIDADAS PARA PACIENTES PKU

29
ENFERMEDADES METABOLICAS
NUESTRA EXPERIENCIA ENFERMEDADES METABOLICAS H.U.M.S

30
SEXO Frecuencia Porcentaje Varón 23 57,5 Mujer 17 42,5 Total 40 100,0
42,5% n= 17 57,5% n=23

31
CLASIFICACIÓN POR EDADES
Frecuencia Porcentaje 0-6 años 9 22,5 >= >= >= >= ,5 >= Total 2 9 11 6 2 10

32
CLASIFICACIÓN P.K.U Frecuencia Porcentaje HPA benigna 12 30,0
PKU leve ,5 PKU moderada ,5 PKU clasica ,0 Total ,0 PKU Clásica 30% 30% PKU Moderada 22,5% 17,5% HPA benigna PKU Leve

33
TIPO DE TRATAMIENTO 52,5% 6 pacientes con KUVAN Menores 4 años
Frecuencia Porcentaje ni dieta ni fármaco ,5 fármaco ,5 solo dieta ,0 Total ,0 6 pacientes con KUVAN Menores 4 años 2 de ellos inicio RN No efectos secundarios 25% 22,5% 52,5%

34
Desarrollo neurocognitivo
Dra. Beatriz Puga González Psicóloga Centro de Crecimiento Andrea Prader Desde 1989 se lleva a cabo en el Centro Andrea Prader el seguimiento del desarrollo psicomotor e intelectual de los niños afectos de Hiperfenilalaninemia Clásica (PKU), detectados por screening neonatal y tratados precozmente, al disponer de los estándares del desarrollo psicomotor e intelectual del niño normal, elaborados en este Centro. En ese punto de inicio se incorporaron niños de distintas edades, aunque posteriormente se han ido controlando ya desde los primeros meses de vida, estudiándolos sistemáticamente hasta los 18 años de edad.
, detectados por screening neonatal y tratados precozmente, al disponer de los estándares del desarrollo psicomotor e intelectual del niño normal, elaborados en este Centro. En ese punto de inicio se incorporaron niños de distintas edades, aunque posteriormente se han ido controlando ya desde los primeros meses de vida, estudiándolos sistemáticamente hasta los 18 años de edad.")
35
Test empleados Escala de Desarrollo de la 1ª Infancia de Brunet-Lézine, de 3 a 24 meses. Escalas McCarthy de Aptitudes y Psicomotricidad para Niños (MSCA), de 3 a 6 años. Escala de Inteligencia de Wechsler para Niños (WISC), de 7 a 15 años. Escala de Inteligencia de Wechsler para Adultos (WAIS), de 16 a 18 años.
, de 3 a 6 años. Escala de Inteligencia de Wechsler para Niños (WISC), de 7 a 15 años. Escala de Inteligencia de Wechsler para Adultos (WAIS), de 16 a 18 años.")
36
Valores de referencia:
ESTUDIO LONGITUDINAL DEL DESARROLLO PSICOMOTOR E INTELECTUAL DEL NIÑO NORMAL, DESDE LOS 3 MESES HASTA LA EDAD ADULTA (Centro Andrea Prader)* Muestra recogida entre Recién nacidos previamente definidos como normales. Controlados cada 3 meses 1er año cada 6 meses 2º año posteriormente una vez al año. Finalización del estudio entre (18 años de edad). Siempre la misma psicóloga, tanto para este estudio como para el relativo a la PKU. * Ferrández Longás A, Baguer L, Labarta JI, Labena C, Mayayo E, Puga B, Rueda C, Ruiz-Echarri M. Longitudinal Study of Normal Spanish Children from Birth to Adulthood. Antropometric, Puberty, Radiological and Intellectual Data. Pediatr Endocrinol Rev 2005; 2 (Suppl 4):
* Muestra recogida entre Recién nacidos previamente definidos como normales. Controlados cada 3 meses 1er año. cada 6 meses 2º año. posteriormente una vez al año. Finalización del estudio entre (18 años de edad). Siempre la misma psicóloga, tanto para este estudio como para el relativo a la PKU. * Ferrández Longás A, Baguer L, Labarta JI, Labena C, Mayayo E, Puga B, Rueda C, Ruiz-Echarri M. Longitudinal Study of Normal Spanish Children from Birth to Adulthood. Antropometric, Puberty, Radiological and Intellectual Data. Pediatr Endocrinol Rev 2005; 2 (Suppl 4):")
37
PACIENTES PKU - DISTRIBUCIÓN POR EDADES
Nº PACIENTES 3 meses 3 8 años 14 6 meses 12 9 años 9 meses 16 10 años 12 meses 18 11 años 11 18 meses 12 años 10 24 meses 13 años 8 3 años 14 años 4 años 17 15 años 6 5 años 16 años 6 años 17 años 2 7 años 18 años n: 26 (16 varones; 10 mujeres) Abril 2011
 Abril")
38
Resultados en la Escala de Brunet-Lèzine, en SDS*
ÁREAS EDAD, meses 3 n:3 6 n:12 9 n:16 12 n:18 18 24 Control Postural 0,5±0,8 -0,1±1,5 -0,4±0,8 -0,4±0,9 -0,9±1,1 -0,7±1,6 Coordinación Oculo-Motriz -0,7±0,5 0,2±0,8 0,2±1,1 0,1±1,1 0,0±0,8 -0,1±0,6 Lenguaje 0,9±0,2 -0,2±1,0 -1,5±0,7 -1,2±1,0 0,0±1,0 -0,2±0,8 Sociabilidad -0,5±0,9 0,2±0,9 -0,8±0,9 -1,2±0,8 -0,7±1,4 CD TOTAL -0,1±0,3 0,1±1,0 -0,6±0,8 -0,6±0,9 -0,5±1,1 *Controles Centro Andrea Prader

39
Cociente de Desarrollo (CD) Pacientes PKU/grupo control, de 3 a 24 meses
X meses n 233 3 290 12 303 16 299 18 269 16 260 18

40
Resultados en las Escalas McCarthty (MSCA), en SDS*
Edad, años 3 n:14 4 n:17 5 6 Verbal 0,0±1,2 0,1±1,0 0,5±1,0 -0,1±1,2 Perceptivo-Manipulativa -0,6±0,8 -0,8±1,0 -0,3±0,6 -0,6±1,0 Numérica -0,1±1,1 -0,4±1,0 -0,6±0,6 -0,5±1,0 GENERAL COGNITIVA -0,2±1,0 -0,3±0,8 -0,1±0,5 -0,5±1,1 Memoria -0,1±1,0 0,0±1,0 0,0±0,7 -0,4±1,1 Motricidad -0,7±0,6 -0,8±0,9 * Controles Centro “Andrea Prader”

41
Cociente de Intelectual (CI) Pacientes PKU/grupo control, de 3 a 6 años
X años n 237 14 237 17 250 14 258 17

42
Resultados en las Escalas de Wechsler (WISC-WAIS), en SDS*
EDAD n CI VERBAL CI MANIPULATIVO CI TOTAL 7 16 -0,3±0,9 -0,6±0,7 -0,5±0,8 8 14 -0,2±0,8 -0,5±0,6 -0,4±0,7 9 12 -0,9±0,5 10 -0,8±0,7 -0,9±0,7 -1,0±0,6 11 -0,8±0,9 -0,8±0,8 -0,6±1,1 -0,7±0,9 13 -0,4±1,2 -0,7±1,1 -1,0±1,1 -0,6±1,4 -0,9±1,2 15 6 -0,7±1,3 -0,8±1,1 0,0±0,9 -0,4±0,8 -0,2±0,7 17 2 0,2±0,0 0,1±1,3 0,1±0,7 18 3 -0,3±0,1 -0,4±0,4 *Controles Centro Andrea Prader

43
Cociente de Intelectual (CI) Pacientes PKU/grupo control, de 7 a 18 años
X años n 248 16 250 14 243 12 233 12 233 11 235 10 217 8 204 8 201 6 189 6 169 2 158 3

44
Evolución del CD/CI de los pacientes PKU
Edad, años n Pacientes Puntuación X SDS Controles 1 18 101,4±8,3 -0,6±,0,9 107,1±8,9 299 2 101,3±6,7 -0,4±0,8 105,0±8,7 260 3 14 100,9±12,4 -0,2±1,0 102,9±12,6 237 4 17 103,2±11,3 -0,3±0,8 106,9±13,2 5 104,6±6,5 -0,1±0,5 105,7±11,8 250 6 103,6±12,9 -0,5±1,1 109,3±12,1 258 7 16 91,4±8,4 -0,5±0,8 96,8±10,7 248 8 93,4±7,7 -0,4±0,7 97,8±10,9 9 12 92,2±5,5 -0,9±0,5 102,0±10,8 243 10 90,8±7,5 -1,0±0,6 102,9±12,3 233 11 98,2±9,9 -0,8±0,8 107,9±12,5 102,5±12,0 -0,7±0,9 112,3±13,5 235 13 101,4±14,1 -0,7±1,1 110,0±13,1 217 102,0±16,2 -0,9±1,2 114,4±13,5 204 15 104,2±16,4 -0,8±1,1 114,4±16,1 201 109,3±6,5 -0,2±0,7 111,0±10,1 189 120,5±7,8 0,1±0,7 119,7±10,9 169 121,0±4,0 -0,4±0,4 125,8±10,8 158

45
Evolución del CD/CI de los pacientes PKU, en SDS*
años 18 18 14 17 14 17 16 14 12 12 11 10 8 8 6 6 2 3 n *Controles Centro Andrea Prader

46
CONCLUSIONES Dado el escaso número de pacientes no se pueden generalizar estos resultados a la población PKU. Sin embargo, los resultados obtenidos tienen el valor propio de un estudio longitudinal y de la comparación con estándares propios obtenidos por la misma persona (error inter-observador cero, e intra-observador mínimo). En conjunto estos pacientes presentan un desarrollo psicomotor e intelectual normal, aunque prácticamente a todas las edades con valores por debajo de la media.
. En conjunto estos pacientes presentan un desarrollo psicomotor e intelectual normal, aunque prácticamente a todas las edades con valores por debajo de la media.")
47
CONCLUSIONES Bien un comienzo prenatal, bien la posibilidad de mantener un equilibrio perfecto entre un aporte mínimo y óptimo de fenilalanina y un nivel adecuado de este aminoácido en plasma, bien los niveles plasmáticos de tirosina, o bien la influencia de otros factores externos a la anomalía, podrían ser los responsables de estos pobres resultados.

48
El tto con sapropterina es una alternativa real al tto dietético.
Patrón similar de aa y ac grasos Lo cual no excluye un control estricto Época de mayor vulnerabilidad del SNC Permite un adecuado seguimiento del paciente. 25 años después hay que replantearse los objetivos y metas clínicas y terapéuticas. El aumento de la supervivencia y la desaparicon de los problemas neurológicos condiciona la aparición de nuevos retos a los que dar respuesta.

49
AGRADECIMIENTOS A.BALDELLOU I.GARCÍA B.PUGA LABORATORIO PACIENTES PKU
Y SUS FAMILIAS

50
GRACIAS

Presentaciones similares







>")














