Descargar la presentación
La descarga está en progreso. Por favor, espere

1
Paraparesia Espástica Familiar
Marcelo Jara Muñoz Dr. Guillermo Fariña Becado Neurología HBLT Neurólogo Infantil Paraparesia Espástica Familiar

2
Definición Las Paraparesias Espásticas Hereditarias son un grupo de enfermedades neurodegenerativas clínica y genéticamente heterogéneas predominantemente caracterizadas por espasticidad progresiva en extremidades inferiores

3
Historia Descrita Inicialmente en 1880
Ernest A. Strümpell y Maurice Lorrain

5
Epidemiologia Prevalencia de 0.004 a 0.01% 2-10 por 100.000 hab.
La prevalencia varia a través del mundo: 18.4 en Guam, 2.0 in Dinamarca, 2.1 En Libia,9.6 en España, 2.7 en Italia Estas diferencias probablemente se deban al origen geográfico de determinados genes Encyclopedia of movement Disorders, 2010, Pages

6
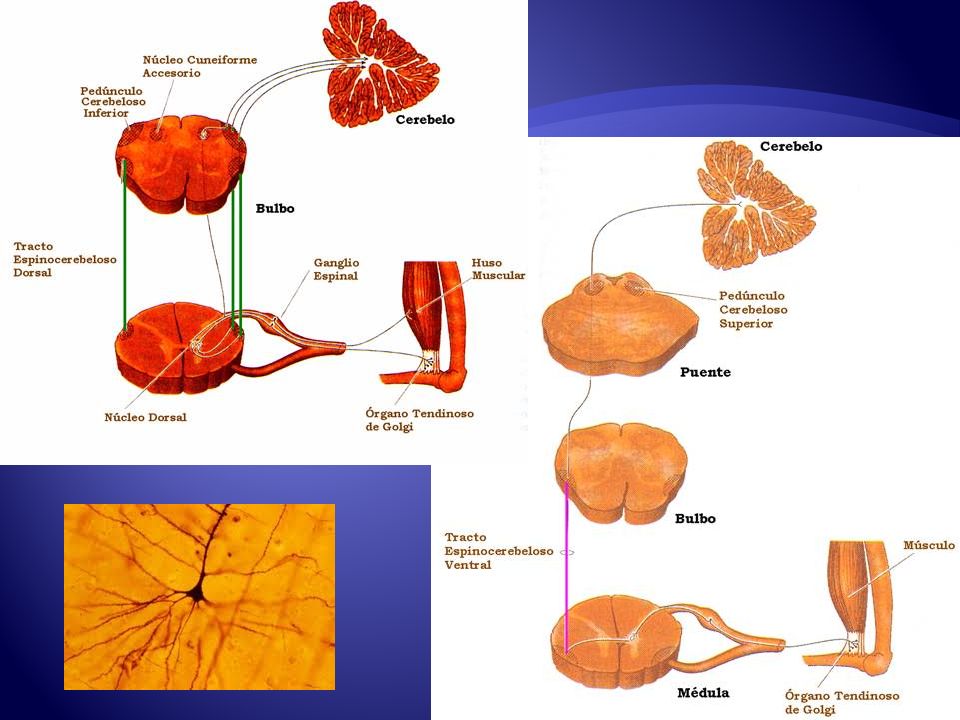
Fisiopatología Estudios neuropatológicos demuestran degeneración axonal de las porciones terminales de las vías ascendentes y descendentes de la medula espinal. Vía cortico-espinal y cordones posteriores N. Lang et al. / Clinical Neurophysiology 122 (2011) 1417–1420
 1417–1420.")
7
Fisiopatología Causado por la alteración de la dinámica de microtúbulos y organelos de transporte y citoesqueleto axonal. Es resultado del largo de los axones ( 1 mt), y que son extremadamente dependiente del trafico axonal. Lancet Neurol 2008; 7: 1127–38
, y que son extremadamente dependiente del trafico axonal. Lancet Neurol 2008; 7: 1127–38.")
8
Lancet Neurol 2008; 7: 1127–38

9
Fisiopatología Se observa además compromiso de vía espinocerebelosa
Hallazgos menos comunes Disminución del numero de células de Betz en capa V de corteza motora Perdida Neuronal en columna de Clark Perdida neuronal en ganglios basales y cerebelo J Neurol Neurosurg Psychiatry :

11
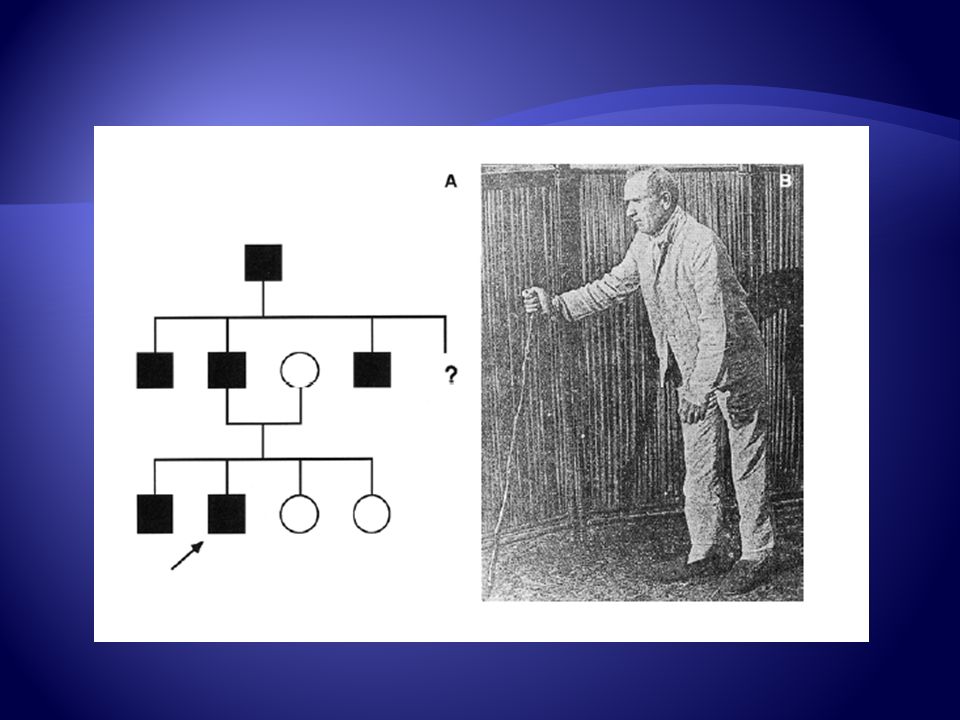
Clasificación Modo de Herencia Autosómica dominante
Autosómica recesiva Ligada a Cromosoma X

12
Clasificación PEF puede clasificarse clínicamente como:
Pura (No Complicada) Compleja (Complicada) Ataxia Cerebelosa, Deterioro Cognitivo, Neuropatía Periférica, Atrofia óptica, otros.
 Compleja (Complicada) Ataxia Cerebelosa, Deterioro Cognitivo, Neuropatía Periférica, Atrofia óptica, otros.")
13
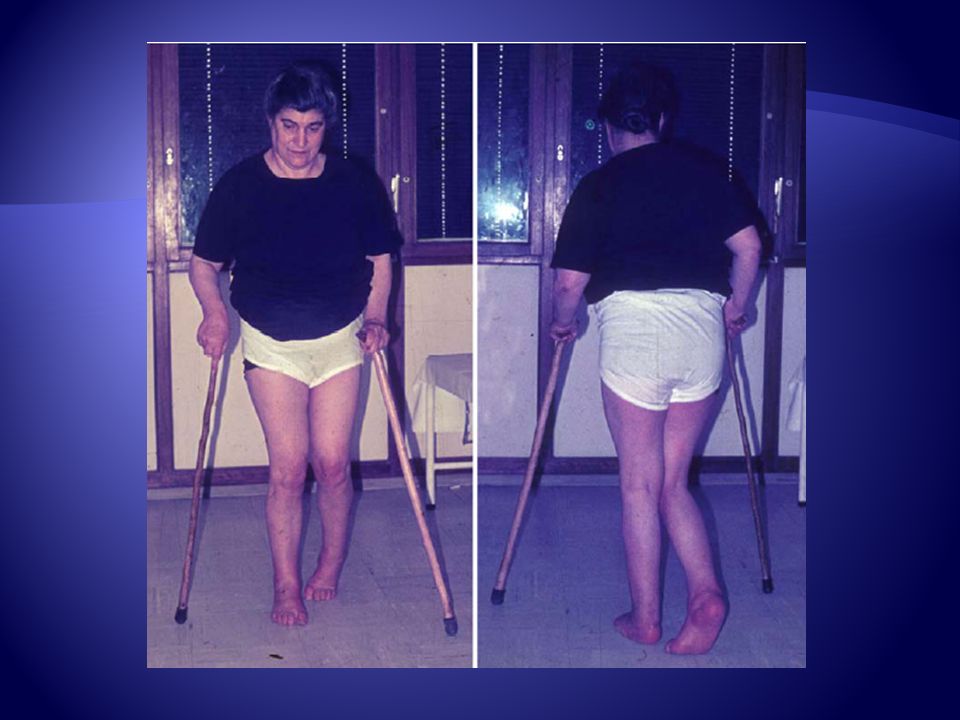
Clínica Inicio sutil y variable, desde primera a séptima década de la vida Es característico relativa preservación de fuerza en relación con aumento del tono en extremidades inferiores, asocia hiperreflexia, clonus y plantares extensores.

14
Clínica Existe preservación de extremidades superiores
Puede asociar síntomas sensitivos Abatiestesia Apalestesia Alteraciones Urinarias

16
Clínica Síntomas, signos de complicación: Ataxia Amiotrofia Severa
Atrofia Óptica Retinopatía pigmentaria Retardo mental Signos extrapiramidales Sordera Ictiosis Neuropatía periférica y epilepsia

21
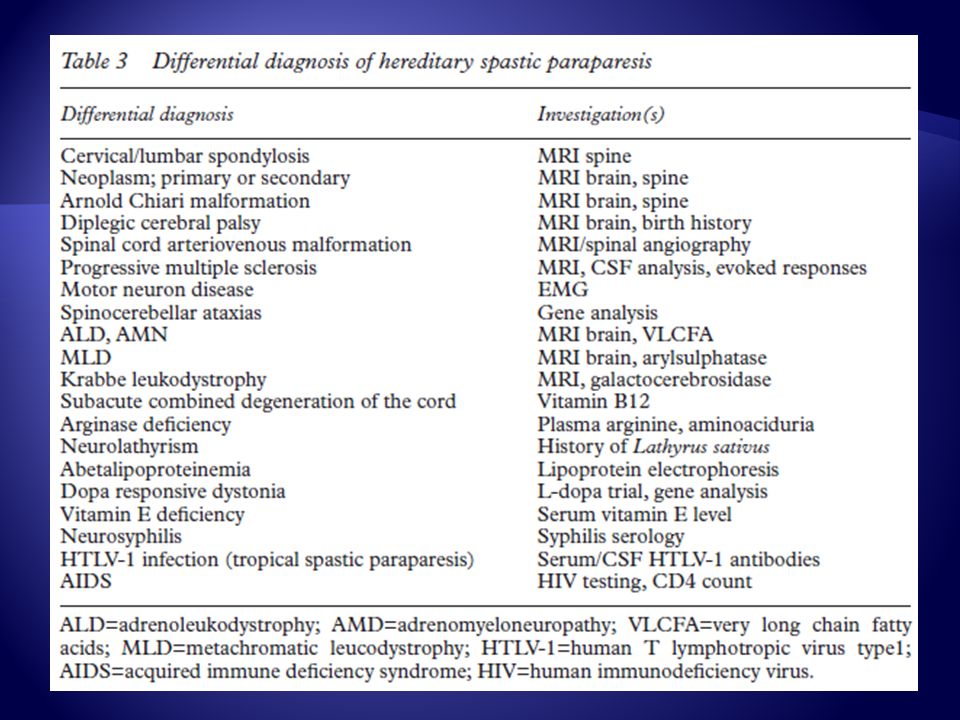
Diagnostico Diferencial
Inicio en la Infancia Parálisis cerebral Diplejica Estructural (Chiari- Subluxación Atlanto Axoidea) Leucodistrofia Distonía Respondedora de L-Dopa Mielitis infecciosa Esclerosis Múltiple
 Leucodistrofia. Distonía Respondedora de L-Dopa. Mielitis infecciosa. Esclerosis Múltiple.")
22
Diagnostico Diferencial
Inicio Adulto: Enfermedad degenerativa columna cervical Esclerosis Múltiple Enfermedad de Motoneurona Neoplasias Mielitis Infecciosas Malformación AV Dural

23
Diagnostico Diferencial
Inicio Adulto: Deficiencia de Vit. (B12-E) Ataxias Espinocerebelosas Latirismo Distonía Respondedora de L-Dopa Infecciones (VDRL-VIH-HTLV1) Deficiencia de Cobre
 Ataxias Espinocerebelosas. Latirismo. Distonía Respondedora de L-Dopa. Infecciones (VDRL-VIH-HTLV1) Deficiencia de Cobre.")
24
Estudio Ácidos grasos de cadena larga Aminoácidos en plasma
Vit. B12 y Vit. E Ceruloplasmina y cobre Serología VDRL, VIH y HTLV1 Evaluación neuro-oftalmológica

25
Estudio Cariograma PEA,PEV y PESS EMG y VCN RM cerebral y medula total
Muestra especifica para Paraparesia Espástica Familiar a estudiar

27
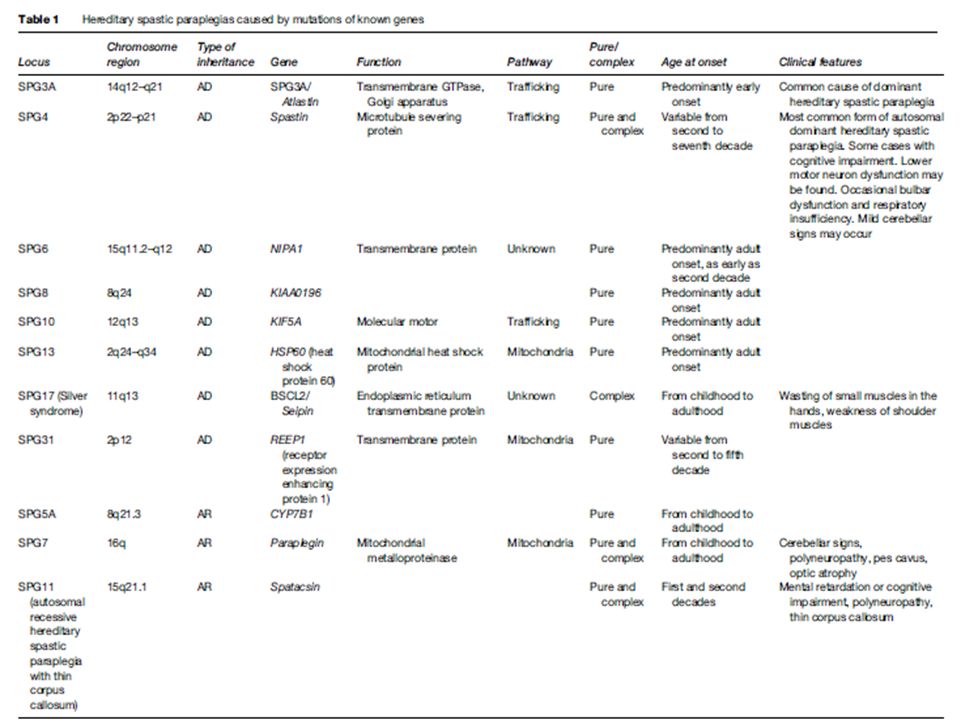
Autosómicas Dominantes
SPG4 (Strumpell–Lorrain) La mas común, % del total de casos de AD Mutación en el gen SPAST cromosoma 2p22, que codifica para la Espastina Involucrada en la red de microtúbulos y transporte axonal Gran variación fenotípica incluso intrafamiliar
 La mas común, % del total de casos de AD. Mutación en el gen SPAST cromosoma 2p22, que codifica para la Espastina. Involucrada en la red de microtúbulos y transporte axonal. Gran variación fenotípica incluso intrafamiliar.")
28
Autosómicas Dominantes
SPG4 (Strumpell–Lorrain) Clínica: Habitualmente considerada de las formas Puras, pero en 50% de los casos, asocia alteraciones esfinterianas, alteraciones de sensibilidad y PNP. Casos aislados de epilepsia, ataxia, RM y adelgazamiento de cuerpo calloso Edad de presentación suele ser tardía, pero puede ser de 1 a 63 años, peacks a los 10 y 30 años J Neurol Neurosurg Psychiatry 2010;81:
 Clínica: Habitualmente considerada de las formas Puras, pero en 50% de los casos, asocia alteraciones esfinterianas, alteraciones de sensibilidad y PNP. Casos aislados de epilepsia, ataxia, RM y adelgazamiento de cuerpo calloso. Edad de presentación suele ser tardía, pero puede ser de 1 a 63 años, peacks a los 10 y 30 años. J Neurol Neurosurg Psychiatry 2010;81:")
29
Autosómicas Dominantes
SPG3A Es debido a la mutación en el gen ATL1 el cual codifica para la Atlastina-1 Atlastina-1 está implicada en facilitar las interacciones de membrana, de fisión y en determinados casos, la fusión. Atlastina interactúa con el retículo endoplasmático de la membrana en neuronas corticoespinal para coordinar, dar forma y dinámica de los microtúbulos

30
Autosómicas Dominantes
SPG3A Responsable del 10% de las PEF AD Clínica: La mayoría, se presenta en forma pura. Inicio, alrededor e los 4 años. Variedad complicada se presenta como PNP SM axonal.

31
Autosómicas Dominantes
SPG6 Mutaciones en el gen NIPA1 en el cromosoma 15q11.2. NIPA1 codifica la proteína NIPA1 que es una unión directa de atlastin-1. NIPA1 es un inhibidor de la proteína morfogénica ósea (PMO) . Otros inhibidores de la señalización de PMO son espastina y espartina. PMO es importante para la función axonal distal. Representan el 3,6% de la AD-SPG en la población china, pero parecen ser poco frecuentes en Europa. Clínica: la mayoría de los casos se presentan en forma "Pura“ . La Espasticidad es grave y rápidamente progresiva. RM puede revelar atrofia de la médula espinal. Inicio: Adolescencia.
 . Otros inhibidores de la señalización de PMO son espastina y espartina. PMO es importante para la función axonal distal. Representan el 3,6% de la AD-SPG en la población china, pero parecen ser poco frecuentes en Europa. Clínica: la mayoría de los casos se presentan en forma Pura . La Espasticidad es grave y rápidamente progresiva. RM puede revelar atrofia de la médula espinal. Inicio: Adolescencia.")
32
Autosómicas Dominantes
SPG9 Etiología: el gen mutado aún no ha sido detectado Clínica: SPG9 representa un SPG complejo con cataratas, reflujo gastroesofágico, y PNP motor, Se presenta desde la niñez hasta la edad adulta.

33
Autosómicas Recesivas
SPG5 Fenotipo Puro, con edad de inicio va desde 1 a 20 años. Debido a mutación de CYP7B1

34
Autosómicas Recesivas
SPG7 Gen codifica para Paraplejina Da cuenta del 5% del total de Autosómicas recesivas Fenotipos Puro y complicado, signos cerebelosos (disartria, nistagmo y ataxia), discos ópticos pálidos, y neuropatía periférica
, discos ópticos pálidos, y neuropatía periférica.")
35
Autosómicas Recesivas
SPG11 Se debe a la mutación en el gen KIAA1840 en el cromosoma 15q el cual codifica para la espatacsina cuya función es desconocida. Espastacsina es expresada principalmente en motoneuronas corticales y espinales con una distribución citoplasmatica. Se co-localiza con proteínas de trafico de vesículas, RE, superficie mitocondrial y microtúbulos e interactúa con miembros complejo AP5 y espastizina (SPG15). SPG11 es la causa mas frecuente de AR-SPG.
. SPG11 es la causa mas frecuente de AR-SPG.")
36
Autosómicas Recesivas
Clínica: Pura y compleja, en variedad compleja presentan deterioro cognitivo, polineuropatía periférica, se pueden agregar nistagmo y compromiso de extremidades superiores y ataxia. Es la variedad que mas se asocia a adelgazamiento del cuerpo calloso. Un subtipo de SPG11 es el síndrome de Kjellin, caracterizado por paraplejia espástica, retardo mental, amiotrofia, y distrofia macular. Inicio en infancia temprana hasta la adultez.

37
Tratamiento Actividad física:
Se debe llevar a cabo una actividad física regular, incluyendo la natación, que ayuda a mantener la potencia muscular y tasa ponderal brindando apoyo psicológico. Las pautas de actividad física pueden ser programadas y supervisadas por un fisioterapeuta.

38
Tratamiento Espasticidad
El componente espástico dinámico es a veces muy intenso predominando en la musculatura aductora de muslos, y flexora plantar con retracción de los tendones de Aquiles. El enfermo compensa la marcha espástica con un cambio postural que implica hiperlordosis lumbar y elevación de la pelvis de la pierna impulsora (signo de Trendelenburg), que a la larga se traduce en lumbalgias y contracturas musculares. EUR J PHYS REHABIL MED 2010;46:423-38
, que a la larga se traduce en lumbalgias y contracturas musculares. EUR J PHYS REHABIL MED 2010;46:")
39
Tratamiento Espasticidad Estiramiento muscular
Fortalecimiento muscular Entrenamiento muscular Biofeedback EUR J PHYS REHABIL MED 2010;46:423-38

40
Tratamiento Espasticidad Terapia de ondas de choque Ultrasonido
Crioterapia Termoterapia Vibración Estimulación Eléctrica EUR J PHYS REHABIL MED 2010;46:423-38

41
Tratamiento Espasticidad:
Con resultados variables, en general pobres, se han empleado fármacos antiespásticos (p.j., baclofeno y dantrolene sódico) y la infiltración de la musculatura hipertónica con toxina botulínica. Probablemente el mejor remedio sea de nuevo un programa de actividad física reglada con estiramiento de la musculatura espasmódica y fortalecimiento de la más debilitada. Neurotherapeutics, Vol. 8, No. 2, 2011
 y la infiltración de la musculatura hipertónica con toxina botulínica. Probablemente el mejor remedio sea de nuevo un programa de actividad física reglada con estiramiento de la musculatura espasmódica y fortalecimiento de la más debilitada. Neurotherapeutics, Vol. 8, No. 2,")
42
Tratamiento Espasticidad
Baclofeno: Agente farmacológico de elección, inhibidor GABA, produce restricción de ingreso de Ca++ presinaptico, lo cual reduce transmisión sinaptica Vida media de 3 a 4 hrs. Efectos Adversos: sedación, fatiga, somnoliento, ataxia, y confusión mental Neurotherapeutics, Vol. 8, No. 2, 2011

43
Tratamiento Espasticidad Baclofeno Intratecal
Administración continua intratecal por bomba. Aumenta efectividad con menos dosis que vo y disminuye sus efectos adversos Indicado en espasticidad intratable Suspensión abrupta: hipertermia maligna Neurotherapeutics, Vol. 8, No. 2, 2011

44
Tratamiento Espasticidad Tiazinadina:
Agonista a2-adrenergico, inhibe la liberación de aminoácidos en la sinapsis de las interneuronas espinales, incrementa inhibición presinaptico. Vida media 2,5 hrs Efectos adversos: sedación, somnolencia, hipotensión, mareos, astenia, debilidad muscular, insomnio, alucinaciones, y fatiga. Efectos adversos son dosis dependientes Neurotherapeutics, Vol. 8, No. 2, 2011

45
Tratamiento Toxina Botulínica
Inhibe liberación de neurotransmisores, por inhibición de exocitosis de vesículas presinapticas, con la subsecuente disminución de liberación de acetilcolina Duración de efecto de 3 a 4 meses Neurotherapeutics, Vol. 8, No. 2, 2011

46
Tratamiento Espasticidad Otras Gabapentina, pregabalina,benzodiazepinas, clonidina, dantroleno, y cannabis Neurotherapeutics, Vol. 8, No. 2, 2011

47
Tratamiento Pie cavo. Como en otros síndromes neurodegenerativos, las deformidades de la arquitectura del pie forma parte del cuadro clínico de PEF. En general se trata de pie cavo-varo, con retracción de los tendones de Aquiles, y deformidad en garra de los dedos de los pies.

48
Tratamiento Pie Cavo La modificación de los arcos plantares agrava las dificultades de la marcha espástica que por definición tiene todo paciente de PEF. El abordaje terapéutico del pie cavo corre a cargo de un equipo multidisciplinario donde intervienen neurólogos, fisioterapeutas, podólogos y traumatólogos.

49
Tratamiento Trastornos urinarios.
Se llega a observar en el 75 a 78% de la PEF ya sean puras o complejas Incontinencia urinaria 69,4% Hesitancy 59,2% Nicturia 55,1% Urgencia 51% J Neurol Neurosurg Psychiatry Mar;81(3): Epub 2009 Sep 2 Spinal Cord Jul;50(7): doi: /sc Epub 2012 Jan 31
: Epub 2009 Sep 2. Spinal Cord Jul;50(7): doi: /sc Epub 2012 Jan 31.")
50
La Evaluación urodinámica demostró:
Signos de vejiga neurogénica central en el 82.7%, con hiperactividad del detrusor en 51.7% y disinergia en el esfínter detrusor 65.5% Residuo postmiccional > al 10% se encontró en un 41,4% Evaluación ultrasonográfica no demostró alteraciones de vía urinaria superior J Neurol Neurosurg Psychiatry Mar;81(3): Epub 2009 Sep 2 Spinal Cord Jul;50(7): doi: /sc Epub 2012 Jan 31
: Epub 2009 Sep 2. Spinal Cord Jul;50(7): doi: /sc Epub 2012 Jan 31.")
51
Tratamiento Trastornos urinarios.
Medidas como el vaciamiento vesical reglado e ingesta programada de líquidos permiten a menudo un aceptable control urinario en las actividades de la vida diaria. Si con esto no es suficiente puede pasarse al tratamiento farmacológico de la hiperrefexia del detrusor con fármacos anticolinérgicos de acción periférica. J Neurol Neurosurg Psychiatry.2010 Mar;81(3): Epub 2009 Sep 2 Spinal Cord Jul;50(7): doi: /sc Epub 2012 Jan 31
: Epub 2009 Sep 2. Spinal Cord Jul;50(7): doi: /sc Epub 2012 Jan 31.")
52
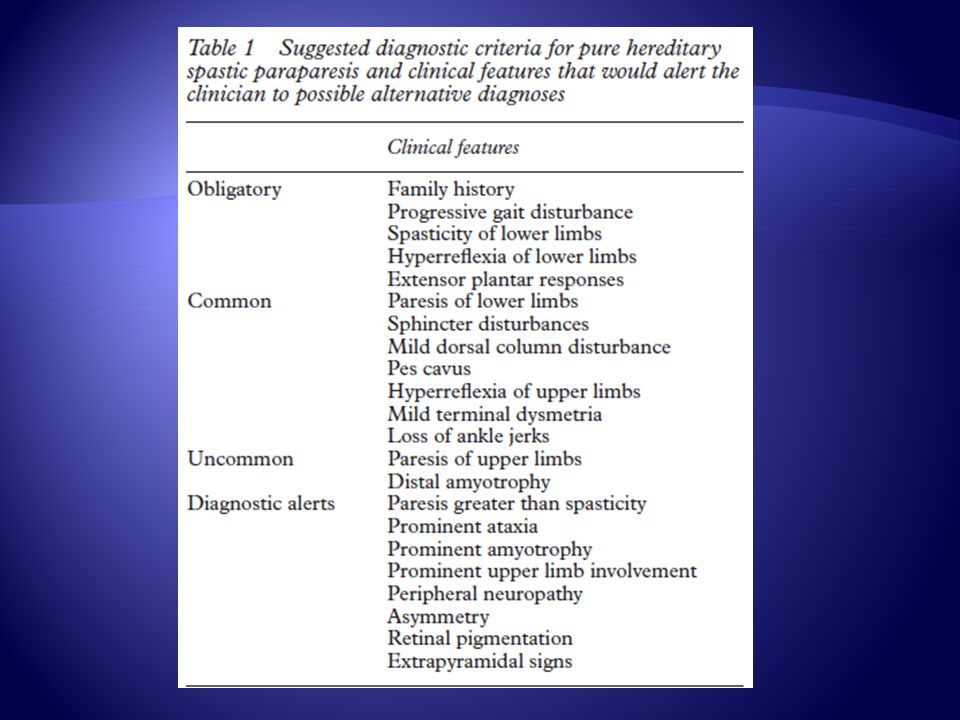
Criterios que sugieren parapesia espástica familiar
Obligatorios Historia familiar Compromiso progresivo Espasticidad Hiperreflexia Plantar extensor Paresia de EEII Compromiso de esfínteres Pie cavo Trastorno de la sensibilidad profunda Hiperreflexia de EESS Dismetría Comunes

53
Infrecuente Signos de Alerta Paresia de extremidades superiores
Amiotrofia distal Paresia > espasticidad Signos extrapiramidales Ataxia prominente Amiotrofia severa Neuropatía periférica Asimetría Retinas pigmentadas Signos de Alerta

54
Considerando el antecedente Familiar de Polineuropatia Periferica Sensitivo Motora Axonal

55
PEF con Polineuropatía
Tipo Paraparesia Herencia Edad Inicio Clínica SPG3A AD Infancia Pura + PNP SPG4 Adultez SPG7 AR Infancia a Adultez PNP + pie cavo + signos cerebelosos + atrofia óptica SPG11 1ra y 2da décadas RM o deterioro cognitivo+PNP+Cuerpo calloso Delgado SPG15 (Sd Kjlling) Infancia a Adultez Maculopatía Pigmentaria +amiotrofia distal +pie cavo+ disartria+ signos cerebelosos+ demencia+ adelgazamiento de cuerpo calloso+ signos extrapiramidales
 Infancia a Adultez. Maculopatía Pigmentaria +amiotrofia distal +pie cavo+ disartria+ signos cerebelosos+ demencia+ adelgazamiento de cuerpo calloso+ signos extrapiramidales.")
56
PEF con Polineuropatía
Tipo Paraparesia Herencia Edad Inicio Clínica SPG21 (Sd de Mast) AR Infancia a Adultez Temprana Prematuros; RDSM; disartria; Signos Cerebelosos; Sd Extrapiramidal; Cuerpo Calloso Delgado; Cataratas; Distonia; Corea SPG9 AD Infancia a Adultez Cataratas; Neuropatia Motora; Talla Baja; Anormalidades Esqueleticas; RGE SPG19 Adultez Lenta y progresión benigna, polineuropatia SPG14 RM, neuropatia distal, Agnosia Visual SPG23 (Sd Lison) Infancia Temprana Anormalidades pigmentarias de piel y pelo; Dismorfias Faciales y Esqueleticas, Signos Cerebelosos
 AR. Infancia a Adultez Temprana. Prematuros; RDSM; disartria; Signos Cerebelosos; Sd Extrapiramidal; Cuerpo Calloso Delgado; Cataratas; Distonia; Corea. SPG9. AD. Infancia a Adultez. Cataratas; Neuropatia Motora; Talla Baja; Anormalidades Esqueleticas; RGE. SPG19. Adultez. Lenta y progresión benigna, polineuropatia. SPG14. RM, neuropatia distal, Agnosia Visual. SPG23. (Sd Lison) Infancia Temprana. Anormalidades pigmentarias de piel y pelo; Dismorfias Faciales y Esqueleticas, Signos Cerebelosos.")
57
PEF con Polineuropatía
Tipo Paraparesia Herencia Edad Inicio Clínica SPG3A AD Infancia Pura + PNP SPG4 Adultez SPG7 AR Infancia a Adultez PNP + pie cavo + signos cerebelosos + atrofia óptica SPG11 1ra y 2da décadas RM o deterioro cognitivo+PNP+Cuerpo calloso Delgado SPG15 (Sd Kjlling) Infancia a Adultez Maculopatía Pigmentaria +amiotrofia distal +pie cavo+ disartria+ signos cerebelosos+ demencia+ adelgazamiento de cuerpo calloso+ signos extrapiramidales
 Infancia a Adultez. Maculopatía Pigmentaria +amiotrofia distal +pie cavo+ disartria+ signos cerebelosos+ demencia+ adelgazamiento de cuerpo calloso+ signos extrapiramidales.")
Presentaciones similares

















, aunque sus síntomas.>")





