Descargar la presentación
La descarga está en progreso. Por favor, espere


1
ENZIMOPATIAS ERITROCITARIAS
Química Biológica Patológica ENZIMOPATIAS ERITROCITARIAS I-Deficiencia G6PD II- Def. Piruvato Quinasa Tema:3 (Bolilla3) Dra. Silvia Varas
 Dra. Silvia Varas.")
2
Programa QBP TEMA 3: Alteraciones en el metabolismo de hemoglobina: Hemoglobinas estructurales y Talasemias, Consecuencias clínicas y metabólicas, diagnóstico bioquímico y molecular. Mecanismo molecular del defecto genético. Enzimopatías eritrocitarias: deficiencia de piruvatoquinasa y glucosa 6-fosfato deshidrogenasa.

3
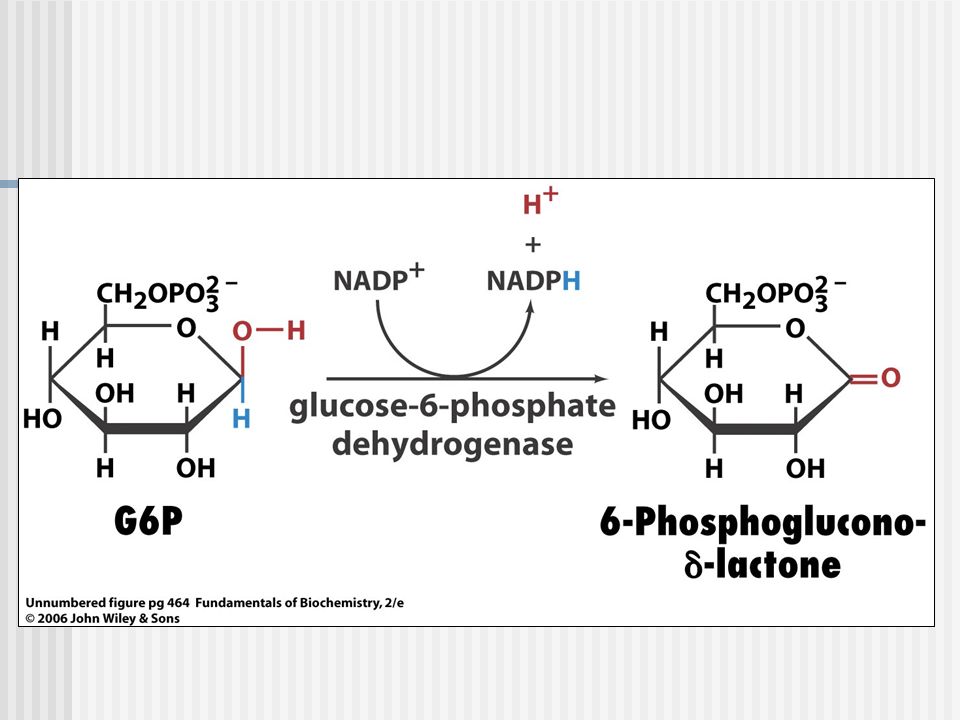
Deficiencia de Glucosa 6 P-deshidrogenasa
Historia

4
Habas (Vicia faba) 1- Teoría Tóxica Pitágoras ( a. C.)
 1- Teoría Tóxica Pitágoras ( a. C.)")
5
Historia 2º- “teoría alérgica” asociado al consumo de las habas, en un principio no se había asociado a una enfermedad heredable. 3º-Una tercera etapa fue el reconocimiento de una anemia hemolítica causado por drogas.

6
Historia La primaquina es una droga usada para el tratamiento de la malaria. Se acuño el termino de “primaquina sensible” a los eritrocitos de los individuos con esta deficiencia

7
El estadio final del descubrimiento por Carson y col
El estadio final del descubrimiento por Carson y col. (1956) quien demostraron que el defecto metabólico primario en sujetos susceptibles a la primaquina esta asociada a una baja actividad de la G6PD en los eritrocitos. Hacia 1958, Gross et al., por un lado y Szeinberg et al. por otro, determinaron que la deficiencia enzimática tenía una base hereditaria y sugirieron que estaba ligada al sexo.
 quien demostraron que el defecto metabólico primario en sujetos susceptibles a la primaquina esta asociada a una baja actividad de la G6PD en los eritrocitos. Hacia 1958, Gross et al., por un lado y Szeinberg et al. por otro, determinaron que la deficiencia enzimática tenía una base hereditaria y sugirieron que estaba ligada al sexo.")
8
Gen G-6PD Hemofilia A Disqueratosis Congénita Ceguera al Color

9
G6PD En los eritrocitos se encuentra en sus formas dimérica y tetramérica y las dos formas estan en un equilibrio dependiente de pH, con iguales proporciones a pH neutro La actividad catalítica sólo se inicia cuando se establece una asociación en estado de equilibrio entre las formas dimérica y tetramérica. Tal asociación requiere de la presencia de NADP+. La disminución del pH provoca un cambio hacia la forma tetramérica Figura Nº2: Un dímero de glucosa-6-fosfato deshidrogenada humano. Subunidades A y B son de color rojo y azul. Las moléculas estructurales de NADP+ se dibujan en un modelo y es de color azul oscuro.

11
Ruta de las Pentosas Fosfato de Oxidación

13
GSSG: glutathione disulfide (oxidized glutathione)
Reacciones en los eritrocitos en las que se produce y es destoxificado de H2O2

15
Formación Cuerpos Heinz
OxiHb (6HS) Formación Cuerpos Heinz NADH NAD+ - Superóxido dismutasa MetaHb (6HS) O2 Hb sin hem Hemicromo 1 (Reversible) Hemicromo 2 (Irreversible) R H2O2 GSH Catalasa Cadenas precipitadas Precipita (4 HS) I Glutation peroxidasa GSSG Cuerpos de Heinz (unido MP) H2O LISIS
 Formación Cuerpos Heinz. NADH. NAD+ - Superóxido dismutasa. MetaHb (6HS) + O2. Hb sin hem. Hemicromo 1 (Reversible) Hemicromo 2 (Irreversible) R. H2O2. GSH. Catalasa. Cadenas precipitadas. Precipita (4 HS) I. Glutation peroxidasa. GSSG. Cuerpos de Heinz (unido MP) H2O. LISIS.")
16
(Anion Exchanger 1)
")
17
Mecanismo Molecular de Lisis
La posible secuencia de eventos seria: Los hemicromos se unen a banda 3, catalizan la oxidación de Banda 3. Entrecruzamiento oxidativo de Banda 3. Inmediata asociación con Syk (Spleen tyrosine kinase), fosforilación de treoninas de Banda 3. La fosforilación marca la disminución de afinidad con la anquirina y liberación del citoesqueleto (espectrina/actina). Aumenta la movilidad lateral en la bicapa (clustering) de Banda 3. Formación de agregados de Banda 3. Progresiva vesiculación. Perdida de la membrana plasmática y LISIS celular
, fosforilación de treoninas de Banda 3. La fosforilación marca la disminución de afinidad con la anquirina y liberación del citoesqueleto (espectrina/actina). Aumenta la movilidad lateral en la bicapa (clustering) de Banda 3. Formación de agregados de Banda 3. Progresiva vesiculación. Perdida de la membrana plasmática y LISIS celular.")
18
Tomado de: Pantaleo A, Ferru E, Carta F, Mannu F, Simula LF, et al
Tomado de: Pantaleo A, Ferru E, Carta F, Mannu F, Simula LF, et al. (2011) Irreversible AE1 Tyrosine Phosphorylation Leads to Membrane Vesiculation in G6PD Deficient Red Cells. PLoS ONE 6(1): e15847
 Irreversible AE1 Tyrosine Phosphorylation Leads to Membrane Vesiculation in G6PD Deficient Red Cells. PLoS ONE 6(1): e")
19
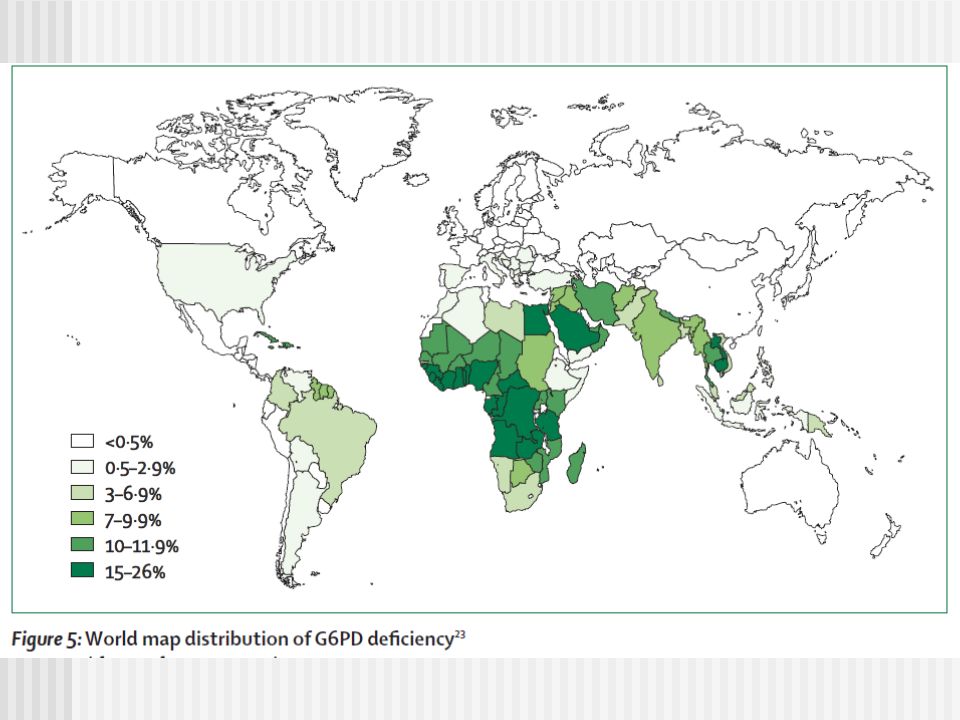
Deficiencia de G6PD G6PD deficiencia es un defecto genético hereditario ligado-X. El defecto es causado por mutaciones en el gen G6PD, que resulta en variantes de proteína con diferentes niveles de actividad enzimática, Esto esta asociado a una amplia rango de fenotipos clínicos y bioquímicos. La prevalencia global es estimada en 4,9%, con alrededor de 330 millones de personas afectados en todo el mundo.

21
Deficiencia de G6PD: Variantes
De acuerdo con su nivel de actividad, las variantes de la enzima, se agruparon en cinco clases, que son: Clase 1: Deficiencia severa de la enzima con anemia hemolítica crónica no esferocítica (CNSHA). Clase 2: Deficiencia enzimática severa (1- 10% actividad residual) asociado con anemia hemolítica aguda. Clase 3: Deficiencia enzimática moderada (10%- 60%, actividad residual). Clase 4: Actividad Normal (60%-150%). Clase 5: Actividad enzimática por encima de lo normal (150%).
. Clase 2: Deficiencia enzimática severa (1- 10% actividad residual) asociado con anemia hemolítica aguda. Clase 3: Deficiencia enzimática moderada (10%- 60%, actividad residual). Clase 4: Actividad Normal (60%-150%). Clase 5: Actividad enzimática por encima de lo normal (150%).")
23
Manifestaciones Clínicas:
1-Anemia Hemolitica inducida por drogas 2-Anemia Hemolitica inducida por infección 3-Favismo 4- Bilirrubina Neonatal

24
1-Anemia Hemolítica inducida por drogas
Son asintomático hasta la administración de un fármaco.

25
1-Anemia Hemolítica inducida por drogas
Cuando ingieren drogas o químicos que desencadenan la hemólisis masiva intravascular El paciente desarrolla fiebre, orina de color negro(Hemoglobinuria), ictericia y anemia La necrosis tubular aguda puede complicar el episodio hemolítico severo En algunos pacientes, la coagulación intravascular diseminada (CID), puede ser una complicación
, ictericia y anemia. La necrosis tubular aguda puede complicar el episodio hemolítico severo. En algunos pacientes, la coagulación intravascular diseminada (CID), puede ser una complicación.")
26
1-Anemia Hemolítica inducida por infección
Virus Hepatitis A y B, citomegalovirus, pneumonia, y fiebre tifoidea son causas comunes Una teoría es que la generación de H2O2 por los PMN neutrófilos puede provocar una disminución en la cantidad de glutatión reducido el estrés oxidativo, por lo que disminuye la capacidad protectora de la célula. La necrosis tubular aguda puede complicar el episodio hemolítico severo En algunos pacientes, la coagulación intravascular diseminada (CID), puede ser una complicación
, puede ser una complicación.")
27
1 y 2-Anemia Hemolítica: CONSECUENCIAS
Fatiga Dolor de Espalda Anemia Ictericia Bilirrubina No Conjugada y Lactato Deshidrogenasa, Y Reticulocitosis son los marcadores del desorden.

28
3-Favismo Consumo de habas frescas
Las sustancias tóxicas asociadas son Divicina, isouramil, y convicina. Manifiesta 24 hs luego de la ingesta Hemoglobinuria es mas severa que la causada por las crisis hemolíticas producidas por drogas o infección (con Bilirrubina baja). Anemia es generalmente aguda y severa, genera falla renal aguda debido a isquemia o a la precipitación de hemoglobina
. Anemia es generalmente aguda y severa, genera falla renal aguda debido a isquemia o a la precipitación de hemoglobina.")
29
4- Bilirrubina Neonatal
Cerca de 1/3 de los neonatos varones nacidos con ictericia neonatal tienen deficiencia de G6PD La ictericia neonatal puede ser subclínica o llevar al kernicterus, que consiste en daño cerebral y de los nervios auditivos por niveles elevados de bilirrubinemia neonatal no conjugada, y puede llevar a discapacidad intelectual, parálisis cerebral, sordera y muerte.

30
Bilirrubina Neonatal BLibre= Bilirrubina Libre/ Bilirrubina Total. 100 = 80%

31
4- Bilirrubina Neonatal
Hacer estudios para Def. G6PD: A los neonatos (1-4d) que desarrollan hiperbilirrubinemia (bilirrubina No Conjugada o Libre (indirecta) concentraciones mayores que 8,8mg/dl [150M/L]) Bilirrubina Indirecta >95%de la Total dentro de las primera 24 h de vida, o en aquellos con antecedentes familiares de hermanos con ictericia
 que desarrollan hiperbilirrubinemia (bilirrubina No Conjugada o Libre (indirecta) concentraciones mayores que 8,8mg/dl [150M/L]) Bilirrubina Indirecta >95%de la Total dentro de las primera 24 h de vida, o en aquellos con antecedentes familiares de hermanos con ictericia.")
32
CARACTERÍSTICAS DE LABORATORIO
Anemia y reticulocitosis Cuerpos de Heinz La presencia de células crenadas o equinocitos

33
Deficiencia de G6PD Control Normal
El Test de cuerpos de Heinz para la sensibilidad de la primaquina (Deficiencia de G6PD). Las células (derecha) son de un donante sensible (G6PD-deficiencia), las células en el panel de la izquierda, de un control normal.
. Las células (derecha) son de un donante sensible (G6PD-deficiencia), las células en el panel de la izquierda, de un control normal.")
34
Una tinción histoquímica indirecta de los eritrocitos de un paciente heterocigoto para la deficiencia de G6PD que sufría de malaria adquirida de forma natural. Las células más pigmentadas son aquellos que contienen actividad normales G6PD. Ellos contienen el mayor porcentaje de parásitos de la malaria, como se muestra por las flechas

35
Diagnóstico Bioquímico: Actividad Enzimática
Medir la actividad enzimática de G6PD en eritrocitos, por métodos cuantitativos con análisis espectro-fotométrico de la tasa de producción de NADPH a partir de NADP+ a 340nm.

36
Diagnóstico Molecular:
Más del 95% de las mutaciones son mutaciones puntuales que generan mutaciones sin sentido o con cambio de sentido. Las mutaciones generan (+) o desaparecen (-) un sitio de corte para una enzima de restricción. Las mutaciones más comunes (Mediterráneo, A-, Seattle, Unión) pueden ser rápidamente detectadas por amplificación del fragmento de interés seguido de digestión con la enzima de restricción (MboII, NlaIII, DdeI o HhaI, respectivamente).
 o desaparecen (-) un sitio de corte para una enzima de restricción. Las mutaciones más comunes (Mediterráneo, A-, Seattle, Unión) pueden ser rápidamente detectadas por amplificación del fragmento de interés seguido de digestión con la enzima de restricción (MboII, NlaIII, DdeI o HhaI, respectivamente).")
37
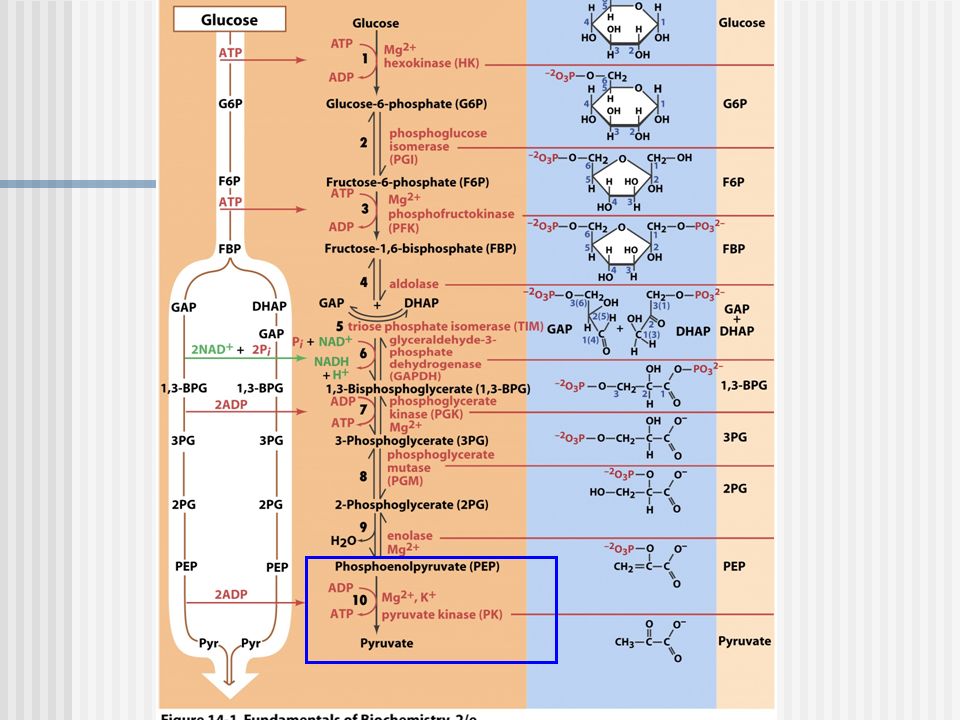
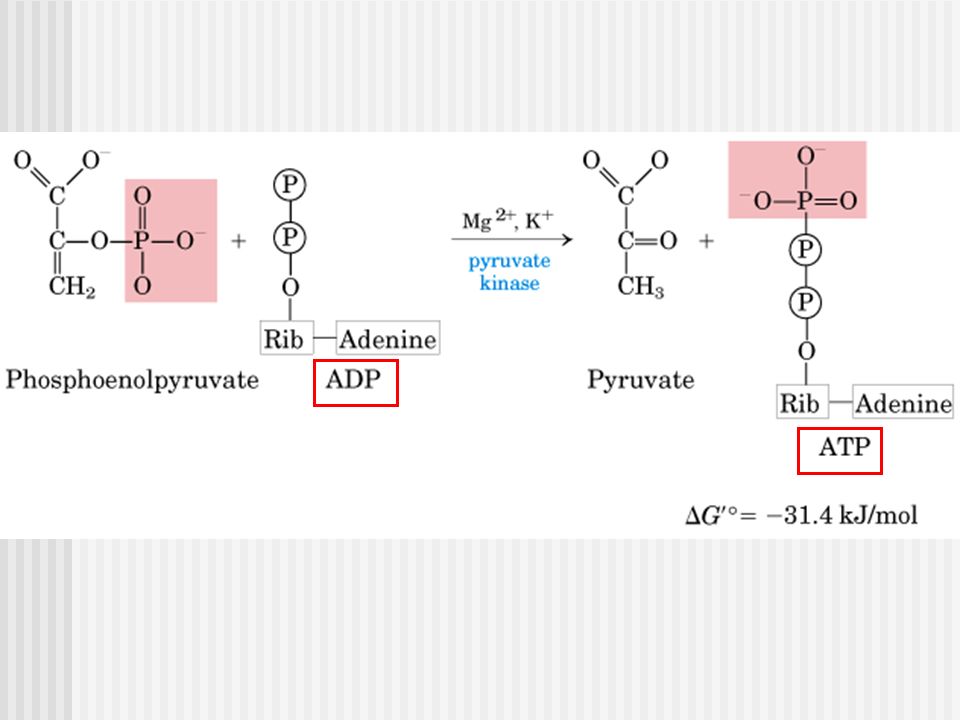
Deficiencia de Piruvato quinasa

40
Deficiencia de PQ PK deficiencia (OMIM 266200) es la causa hereditaria más común de anemia hemolítica no esferocítica (HNSHA) La enfermedad se hereda de forma autosómica recesiva Muestra una distribución en todo el mundo. La prevalencia estimada es de 51 casos por millón (es decir, 1:20.000) en la población blanca.
 en la población blanca.")
41
Piruvato Quinasa Deficiencia
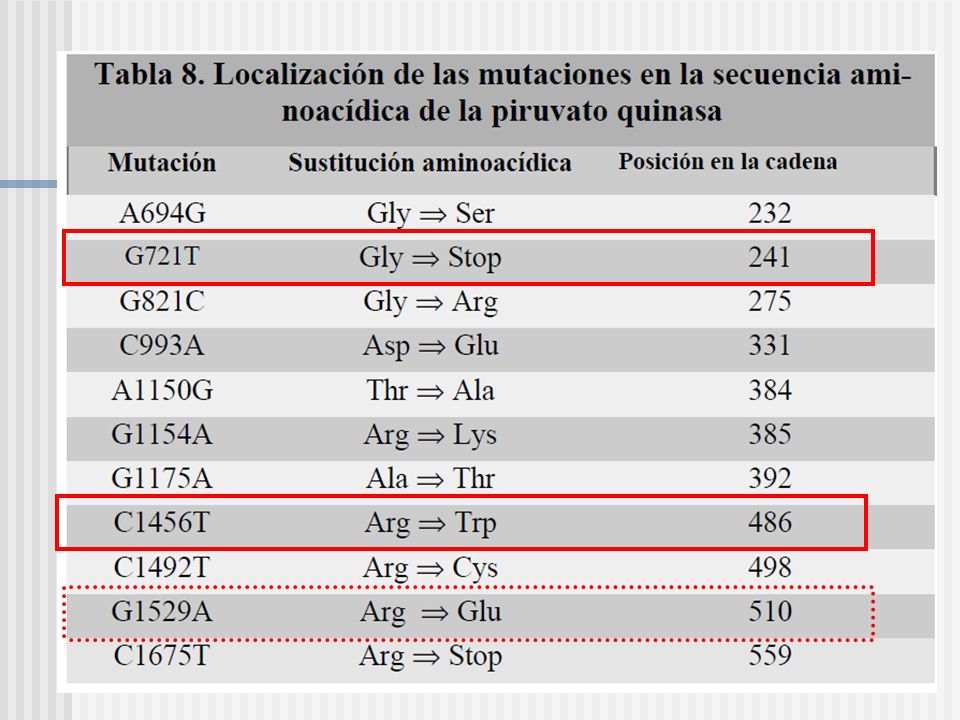
La mutación 1529G A es la mas común en USA y en centro y norte de Europa; La mutación 1456C T es la que prevalece en el sudeste de Europa (España,Italia, Grecia); Y la mutación 1468C T es la mas común en Asia.
; Y la mutación 1468C T es la mas común en Asia.")
43
Características Clínicas
Anemia hemolítica crónica variando en severidad desde asintomática y totalmente compensada a casos con requerimientos de trasfusiones a lo largo de la vida. Grados variables de ictericia suave a moderada esplenomegalia Elevadas concentraciones de 2,3-DPG Otros efectos clínicos incluyen retraso del crecimiento, kernicterus, hidropesía fetal, ulceras crónicas en las piernas, pancreatitis aguda secundaria a enfermedad del tracto biliar, sobrecarga de hierro, absceso esplénico, compresión del cordón espinal por tejido hematopoyético extramedular y flebitis migratoria con trombosis arterial esta son raras complicaciones.

45
Los bajos niveles de hemoglobina pueden ser tolerados debido a una disminución de la afinidad del oxigeno inducido por elevadas concentraciones de 2,3-DPG, característica de deficiencia de PK

46
Hallazgos Hematológicos
Equinocitos En pacientes con esplenectomía se pueden observar siderocitos, células target, y cuerpos de Howell-Jolly La formación de cuerpos de Heinz incubados puede estar incrementada. Al ser una anemia hemolítica, se observa una disminución en la concentración de haptoglobina e hiperbilirrubinemia indirecta e incremento en la excreción de urobilinógeno fecal. Tinción de células con Azul dePrusia

47
Equinocitos (célula crenada)
")
48
Diagnóstico Disminución de la actividad de PK en los eritrocitos.
Los pacientes homocigotas para PK deficiencia la actividad es de un 5-25% de la observada en un eritrocito normal y de un % en portadores heterocigotos.

49
Bioquímica y Biología Molecular: Isoenzimas Naturales
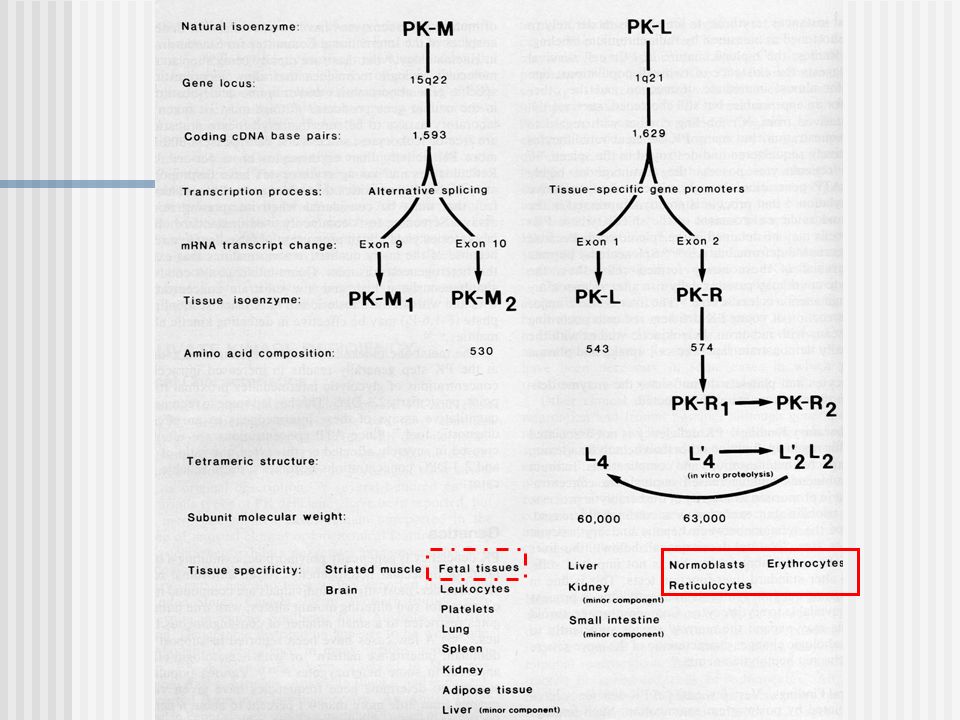
Las isoenzimas relacionadas a un tejido son generalmente homotetrámeros de subunidades de 50 a 60 kDa, productos de genes localizados sobre el cromosoma 1 y 15.

50
PK-M2: determinado en el locus 15q22, puede ser considerado la isoenzima natural prototipo, es la forma presente en varios tejidos durante la vida fetal temprana. Pero PK-M2 persiste como la forma principal en leucocitos, plaquetas, pulmón, riñón, bazo, tejido adiposo y como un componente menor en hígado. PK-M1: es un producto del mismo locus genético obtenido por splicing alternativo y es la forma predominante en músculo estriado maduro y cerebro. Es la única isoenzima que no esta sometida a modulación por sustrato o cofactor. PK-L: es la isoenzima natural predominante en hepatocitos esta codificada por un gen en el cromosoma 1q21. Es un homotetrámero de subunidades L, cada uno constituido por 543 aminoácidos. PK-R: es la tercera isoenzima natural, es única en los eritrocitos maduros. Esta bajo control del mismo locus de PK-L, pero es un producto de transcripción distinto con un extremo 5’ distinto, producto de un promotor especifico de tejido

52
(-)
")
53
RESUMEN:

54
Terapia: El tratamiento será sintomático: Transfusiones de eritrocitos cuando los niveles de Hemoglobina han descendido demasiado. Esplecnotomía en niños con severa anemia, con ello incrementa 1-3 g/dl los niveles de hemoglobina reduciendo los requerimientos de trasfusiones

55
Alguna pregunta?

56
Bibliografía general:
LEHNINGER ALBERT L., COX MICHAEL M., NELSON DAVID L. “Principios de Bioquímica”. Editorial OMEGA 4º Edición Diapositivas en Power Point (formato ppt) (Manual para docentes) LEHNINGER ALBERT L., COX MICHAEL M., NELSON DAVID L. “Principios de Bioquímica” Web: The Journal of Biological Chemistry: Classic Articles Web: VOET DONALD, VOET JUDITH G. “Bioquímica” Editorial MÉDICA PANAMERICANA. 3º Edición, en Español. 2006 WATSON, BAKER, BELL, GANN, LEVINE, LOSICK “Biología Molecular del Gen” Editorial MÉDICA PANAMERICANA. 5º Edición, en Español. 2006 STRYER LUBERT, BERG JEREMY M., TYMOCZKO JOHN L. ”Bioquímica” Editorial REVERTE. Edición 5º Edición, en Español. 2003 MATHEWS CHRISTOPHER K., AHERN KEVIN G., VAN HOLDE K. E. ”Bioquímica” Editorial PEARSON EDUCACION. 3º Edición en Español. 2003 Diapositivas (formato ppt), Problemas (Manual para docentes) y Casos Clínicos de Aplicación VOET DONALD, VOET JUDITH G. and PRATT CHARLOTTE W. “Fundamentals of Biochemistry” Second Edition. Copyright © 2006 by John Wiley & Sons, Inc Web: www. medicapanamericana.com/voet/
 (Manual para docentes) LEHNINGER ALBERT L., COX MICHAEL M., NELSON DAVID L. Principios de Bioquímica Web: The Journal of Biological Chemistry: Classic Articles. Web: VOET DONALD, VOET JUDITH G. Bioquímica Editorial MÉDICA PANAMERICANA. 3º Edición, en Español WATSON, BAKER, BELL, GANN, LEVINE, LOSICK. Biología Molecular del Gen Editorial MÉDICA PANAMERICANA. 5º Edición, en Español STRYER LUBERT, BERG JEREMY M., TYMOCZKO JOHN L. Bioquímica Editorial REVERTE. Edición 5º Edición, en Español MATHEWS CHRISTOPHER K., AHERN KEVIN G., VAN HOLDE K. E. Bioquímica Editorial PEARSON EDUCACION. 3º Edición en Español Diapositivas (formato ppt), Problemas (Manual para docentes) y Casos Clínicos de Aplicación. VOET DONALD, VOET JUDITH G. and PRATT CHARLOTTE W. Fundamentals of Biochemistry Second Edition. Copyright © 2006 by John Wiley & Sons, Inc. Web: www. medicapanamericana.com/voet/")
57
Bibliografía especifica:
- CHARLES R. SCRIVER, ARTHUR L. BEAUDET, WILLIAM S. SLY AND DAVID VALLE: THE METABOLIC AND MOLECULAR BASES OF INHERITED DISEASE. Volume I,II and III. Seventh Edition.Mc Graw-Hill Editors - LUCIO LUZZATTO & ATUL MEHTA. Chapter 111: Glucose 6-Phosphate Dehydrogenase Deficiency. The Metabolic and Molecular Bases of Inherited Diseases. Volume III. Charles R. Scriver, Arthur L. Beaudet, William S. Sly and David Valle, EDITORS. McGraw-Hill, New York. Seventh Edition. (p ). - KOIUCHI R. TANAKA & DONALD E. PAGLIA. Chapter 114: Pyruvate kinase and other enzymopathies of the erythrocyte. The Metabolic and Molecular Bases of Inherited Diseases. Volume III. Charles R. Scriver, Arthur L. Beaudet, William S. Sly and David Valle, EDITORS. McGraw-Hill, New York. Seventh Edition. (p ). - ERNEST BEUTLER: Glucose-6-phosphate dehydrogenase deficiency: a historical perspective. Blood. 2008; 111: - ERNEST BEUTLER: Deficiency G6PD. Blood Vol 84, No 11, 1994 pp - LUCIO LUZZATTO, ATUL MEHTA, TOM VULLIAMY. PART 19: BLOOD: Chapter 179: Glucose 6-Phosphate Dehydrogenase Deficiency. En Scriver CR, Beaudet AL, Sly W S, Valle D (eds). The online metabolic & molecular bases of inherited disease. New York, McGraw Hill, 2012; pag - AKIRA HIRONO, HITOSHI KANNO, SHIRO MIWA, ERNEST BEUTLER: PART 19: BLOOD: Chapter 182: Pyruvate Kinase Deficiency and Other Enzymopathies of the Erythrocyte. En Scriver CR, Beaudet AL, Sly W S, Valle D (eds). The online metabolic & molecular bases of inherited disease. New York, McGraw Hill, 2012; pag - ALBERTO ZANELLA, ELISA FERMO, PAOLA BIANCHI, AND GIOVANNA VALENTINI. Red cell pyruvate kinase deficiency: molecular and clinical aspects. British Journal of Haematology, 2005, 130, 11–25 - AMANDO GARRIDO PERTIERRA (Tesis Doctoral) Genética molecular de la deficiencia eritrocitaria humana en piruvato quinasa. Universidad Complutense de Madrid. España. BLOG:
. - KOIUCHI R. TANAKA & DONALD E. PAGLIA. Chapter 114: Pyruvate kinase and other enzymopathies of the erythrocyte. The Metabolic and Molecular Bases of Inherited Diseases. Volume III. Charles R. Scriver, Arthur L. Beaudet, William S. Sly and David Valle, EDITORS. McGraw-Hill, New York. Seventh Edition. (p ). - ERNEST BEUTLER: Glucose-6-phosphate dehydrogenase deficiency: a historical perspective. Blood. 2008; 111: ERNEST BEUTLER: Deficiency G6PD. Blood Vol 84, No 11, 1994 pp LUCIO LUZZATTO, ATUL MEHTA, TOM VULLIAMY. PART 19: BLOOD: Chapter 179: Glucose 6-Phosphate Dehydrogenase Deficiency. En Scriver CR, Beaudet AL, Sly W S, Valle D (eds). The online metabolic & molecular bases of inherited disease. New York, McGraw Hill, 2012; pag AKIRA HIRONO, HITOSHI KANNO, SHIRO MIWA, ERNEST BEUTLER: PART 19: BLOOD: Chapter 182: Pyruvate Kinase Deficiency and Other Enzymopathies of the Erythrocyte. En Scriver CR, Beaudet AL, Sly W S, Valle D (eds). The online metabolic & molecular bases of inherited disease. New York, McGraw Hill, 2012; pag ALBERTO ZANELLA, ELISA FERMO, PAOLA BIANCHI, AND GIOVANNA VALENTINI. Red cell pyruvate kinase deficiency: molecular and clinical aspects. British Journal of Haematology, 2005, 130, 11–25. - AMANDO GARRIDO PERTIERRA (Tesis Doctoral) Genética molecular de la deficiencia eritrocitaria humana en piruvato quinasa. Universidad Complutense de Madrid. España. BLOG:")
Presentaciones similares





>")














 es una alteración fisiopatólogica sistémica, trombohemorrágica, que se presenta en algunas situaciones.>")


