Descargar la presentación
La descarga está en progreso. Por favor, espere


1
ONCOHEMATOLOGÍA GRANADA PREMIR DOSMIL10
Paloma García Martín Servicio de Hematología y Hemoterapia “Hospital Virgen de las Nieves” GRANADA

2
¿CÉLULA PATOLÓGICA? MORFOLOGÍA CITOQUÍMICA INMUNOFENOTIPO CITOGENÉTICA

4
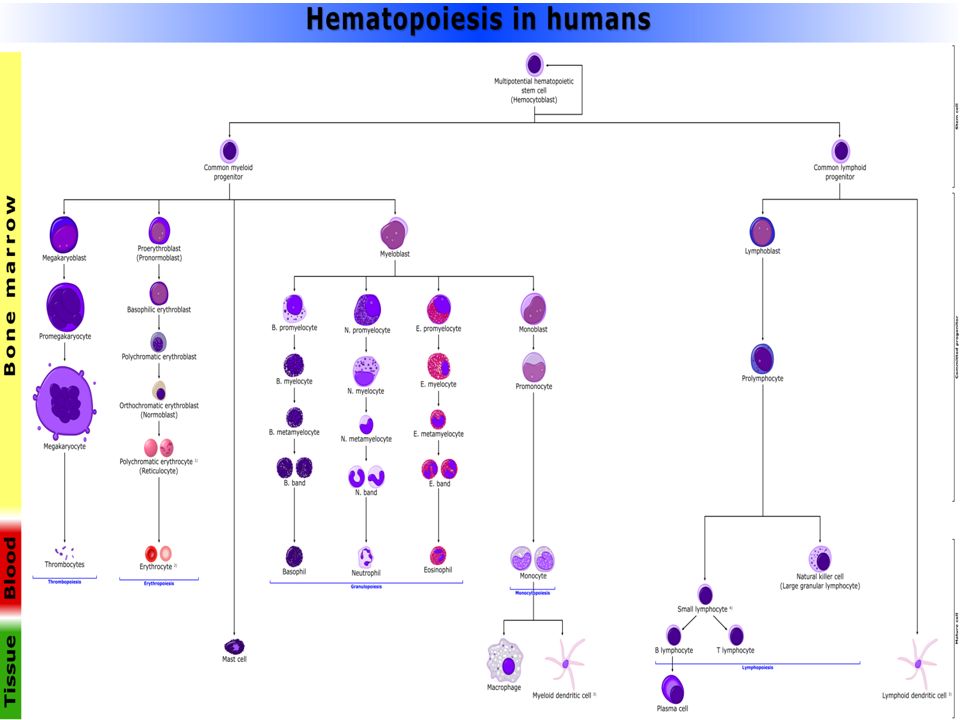
Hematopoyesis LINFOCITOS MÉDULA ÓSEA GANGLIOS LINFÁTICOS

5
Derivan de la MÉDULA ÓSEA
Clínica: EXPRESIÓN EN SANGRE PERIFÉRICA Diagnóstico: BIOPSIA MÉDULA ÓSEA LEUCEMIA AGUDA SÍNDROMES MIELOPROLIFERATIVOS CRÓNICOS GAMMAPATÍAS MONOCLONALES LLA (*) LANL/LMA LMC Policitemia Vera Trombocitemia Esencial Mielofibrosis idiopática Mieloma Múltiple Macroglobulinemia de Waldestron MGUS Enf. de las cadenas pesadas
 LANL/LMA. LMC. Policitemia Vera. Trombocitemia Esencial. Mielofibrosis idiopática. Mieloma Múltiple. Macroglobulinemia de Waldestron. MGUS. Enf. de las cadenas pesadas.")
6
Derivan de los GÁNGLIOS LINFÁTICOS
Clínica: ADENOPATÍAS (*) Diagnóstico: BIOPSIA del GANGLIO LINFOMAS SÍNDROMES LINFOPROLIFERATIVOS CRÓNICOS Linfoma Hodgkin Linfoma Linfocítico (No Hodgkin) LLC-B LLC-T Leucemia/linfoma T del adulto Leucemia Prolinfocítica Síndrome Sezary Tricoleucemia
 Diagnóstico: BIOPSIA del GANGLIO. LINFOMAS. SÍNDROMES LINFOPROLIFERATIVOS CRÓNICOS. Linfoma Hodgkin. Linfoma Linfocítico (No Hodgkin) LLC-B. LLC-T. Leucemia/linfoma T del adulto. Leucemia Prolinfocítica. Síndrome Sezary. Tricoleucemia.")
7
Derivan de los GÁNGLIOS LINFÁTICOS
Clínica: ADENOPATÍAS (*) Diagnóstico: BIOPSIA del GANGLIO LINFOMAS SÍNDROMES LINFOPROLIFERATIVOS CRÓNICOS Linfoma Hodgkin Linfoma Linfocítico (No Hodgkin) LLC-B LLC-T Leucemia/linfoma T del adulto Leucemia Prolinfocítica Síndrome Sezary Tricoleucemia
 Diagnóstico: BIOPSIA del GANGLIO. LINFOMAS. SÍNDROMES LINFOPROLIFERATIVOS CRÓNICOS. Linfoma Hodgkin. Linfoma Linfocítico (No Hodgkin) LLC-B. LLC-T. Leucemia/linfoma T del adulto. Leucemia Prolinfocítica. Síndrome Sezary. Tricoleucemia.")
8
1.a. Linfoma Hodgkin. AP y CLÍNICA
CÉL. REED - STERNBERG CD30 CD25 CD15 Patognomónica Adenopatías Centrípetas Indoloras Signo HOSTER Lesiones óseas osteoblásticas Síntomas B Síndrome VCS Lesión Renal

9
PREDOMINIO LINFOCÍTICO DEPLECCIÓN LINFOCÍTICA
1.a. Linfoma Hodgkin. CLASIFICACIÓN PREDOMINIO LINFOCÍTICO - Niños varones - Linfocitos pequeños - Adenopatía única - MEJOR pronóstico ESCLEROSIS NODULAR** - Mujeres jóvenes - Cél. Lacunares. Fibrosis - A+Masa mediastínica - MÁS frecuente CELULARIDAD MIXTA* - Edad media - Cél neoplásicas=reactivas - Síntomas sistémicos - Relación > con VEB DEPLECCIÓN LINFOCÍTICA - Edad avanzada - Cél RS y focos necrosis - SíntomasB y diseminación - PEOR pronóstico

10
LINFOMA NO HODGKING SUBTIPO HISTOLÓGICO
1.a. Linfoma Hodgkin. ESTADIAJE I Un sólo Grupo Ganglionar o una sola región Extraganglionar (Ie) II Varios grupos ganglionares, al mismo lado del diafragma. Más de un territorio extralinfático por contigüidad (IIe) III Ambos lados del diafragma III1 ½ superior del abdomen (ganglios portales, celíacos y bazo) III2 ½ inferior del abdomen (ganglios paraaórticos, ilíacos y mesentéricos) IIIs Si se afecta el Bazo (III1s, III2s) IIIe Afectación extralinfática por contigüidad IV Diseminado a órganos extralinfáticos con/sin ganglios (sangre y mo) PRONÓSTICO LINFOMA NO HODGKING SUBTIPO HISTOLÓGICO X Masa Bulki (tto RT) A No síntomas B B Síntomas B
 II. Varios grupos ganglionares, al mismo lado del diafragma. Más de un territorio extralinfático por contigüidad (IIe) III. Ambos lados del diafragma. III1. ½ superior del abdomen. (ganglios portales, celíacos y bazo) III2. ½ inferior del abdomen. (ganglios paraaórticos, ilíacos y mesentéricos) IIIs. Si se afecta el Bazo (III1s, III2s) IIIe. Afectación extralinfática por contigüidad. IV. Diseminado a órganos extralinfáticos con/sin ganglios (sangre y mo) PRONÓSTICO. LINFOMA NO HODGKING SUBTIPO HISTOLÓGICO. X Masa Bulki (tto RT) A No síntomas B. B Síntomas B.")
11
ABVD 1.a. Linfoma Hodgkin. TRATAMIENTO RT MOPP ADRIAMICINA BLEOMICINA
VINBLASTINA DACARVACINA Estadío IA ABVD RT MOPP

12
1.b. Linfoma No Hodgkin. CLÍNICA y DG
LINFADENOPATÍAS indoloras y simétricas SÍNTOMAS B: fiebre, sudoración nocturna y pérdida de peso Prurito (No síntoma B) Esplenomegalia LDH (mal pronóstico) TAC BIOPSIA de MO PET
 Esplenomegalia. LDH (mal pronóstico) TAC. BIOPSIA de MO. PET.")
13
LINFOMA HODGKING ESTADÍO DE LA ENFERMEDAD
B 1.b. L. No Hodgkin. CLASIFICACIÓN Leucemia/linfoma linfoblástico de precursores B Linfoma linfocítico de células pequeñas /leucemia linfoma B Trisomía 12. CD5 Menor malignidad ¿†? Síndrome de Richter Linfoma linfoplasmocítico o Inmunocitoma Virus Hepatitis C Linfoma de células del manto t(11;14) bcl-1 CD5 Ciclina D1 Mal pronóstico Gran esplenomegalia Poliposis linfomatoide Linfoma Folicular t(14;18) bcl-2 Cél del centro germinal Linfoma MALT Gástrico Helicobacter Gl. Salivares S. Sjögren Tiroideo T. Hashimoto Linfoma de la zona marginal esplénica Linfoma difuso de células grandes bcl-2 bcl-6 Linfoma primario de cavidades VH 8; + frec VIH Linfoma de Burkitt t(8;14) c-myc Más Maligno “Cielo estrellado” VEB PRONÓSTICO 2º LINFOMA HODGKING ESTADÍO DE LA ENFERMEDAD 1º
 bcl-1 CD5. Ciclina D1. Mal pronóstico. Gran esplenomegalia. Poliposis linfomatoide. Linfoma Folicular. t(14;18) bcl-2. Cél del centro germinal. Linfoma MALT. Gástrico Helicobacter. Gl. Salivares S. Sjögren. Tiroideo T. Hashimoto. Linfoma de la zona marginal esplénica. Linfoma difuso de células grandes. bcl-2 bcl-6. Linfoma primario de cavidades. VH 8; + frec VIH. Linfoma de Burkitt. t(8;14) c-myc. Más Maligno. Cielo estrellado VEB. PRONÓSTICO. 2º. LINFOMA HODGKING ESTADÍO DE LA ENFERMEDAD. 1º.")
14
IPI 1.b. Linfoma No Hodgkin. PRONÓSTICO EDAD > 60 ESTADÍO III Y IV
EXTRAGANGLIONAR MEG (ECOG) LDH FACT RIESGO 0-1 Bajo 2 Bajo-intermedio 3 Intermedio-alto 4 Alto
 LDH FACT. RIESGO Bajo. 2. Bajo-intermedio. 3. Intermedio-alto. 4. Alto.")
15
1.b. L. No Hodgkin. TRATAMIENTO
CHOP Ciclofosfamida Adriamicina Vincristina Prednisona Rituximab Ac Monoclonal anti CD20

16
Derivan de los GÁNGLIOS LINFÁTICOS
Clínica: ADENOPATÍAS (*) Diagnóstico: BIOPSIA del GANGLIO LINFOMAS SÍNDROMES LINFOPROLIFERATIVOS CRÓNICOS Linfoma Hodgkin Linfoma Linfocítico (No Hodgkin) LLC-B LLC-T Leucemia/linfoma T del adulto Leucemia Prolinfocítica Síndrome Sezary Tricoleucemia
 Diagnóstico: BIOPSIA del GANGLIO. LINFOMAS. SÍNDROMES LINFOPROLIFERATIVOS CRÓNICOS. Linfoma Hodgkin. Linfoma Linfocítico (No Hodgkin) LLC-B. LLC-T. Leucemia/linfoma T del adulto. Leucemia Prolinfocítica. Síndrome Sezary. Tricoleucemia.")
17
Infecciones y 2ª Neoplasia Anemia y Trombocitopenia
2.a. LLC-B** Autoinmunidad Infecciones y 2ª Neoplasia Anemia y Trombocitopenia Asintomático CD19 CD20 CD5 Trisomía 12 PRONÓSTICO Evolución a Leucemia Prolinfocítica Síndrome de Richter: posible transformación Linfoma Inmunoblástico tamaño de ganglios y bazo LDH TRATAMIENTO Se inicia si: Síntomas Linfadenopatías deformantes Leucocitosis > FLUDARABINA CICLOFOSFAMIDA RITUXIMAB

18
2.c. Leucemia/Linfoma T del Adulto
2.b. LLC-T 2.c. Leucemia/Linfoma T del Adulto H T L V I ESTRONGILOIDES 2.d. Leucemia Prolinfocítica Mala evolución de una LLC CD5 negativo Mucha más leucocitosis Esplenomegalia >>>

19
2.e. Síndrome de Sezary 2.f. Tricoleucemia M F I U C N O G S O I I S D
Biopsia Piel Cél CD4 Núcleo Cerebriforme Microabscesos de Pautrier Invasión medular 2.f. Tricoleucemia CLÍNICA: Pancitopenia Esplenomegalia dolorosa Infecciones Asocia vasculitis Adenopatías retroperiotneales DIAGNÓSTICO Biopsia de mo TRATAMIENTO Cladribina Rituximab Esplenectomía * Cél B con prolongaciones. Fosfatasa ácida tartrato resistente. CD25 +

20
Derivan de la MÉDULA ÓSEA
Clínica: EXPRESIÓN EN SANGRE PERIFÉRICA Diagnóstico: BIOPSIA MÉDULA ÓSEA LEUCEMIA AGUDA SÍNDROMES MIELOPROLIFERATIVOS CRÓNICOS GAMMAPATÍAS MONOCLONALES LLA (*) LANL/LMA LMC Policitemia Vera Trombocitemia Esencial Mielofibrosis idiopática Mieloma Múltiple Macroglobulinemia de Waldestron MGUS Enf. de las cadenas pesadas
 LANL/LMA. LMC. Policitemia Vera. Trombocitemia Esencial. Mielofibrosis idiopática. Mieloma Múltiple. Macroglobulinemia de Waldestron. MGUS. Enf. de las cadenas pesadas.")
21
1. Leucemias Agudas

22
LMA / LANL LAL

23
1.a. L. Aguda Linfoblástica. CLÍNICA
Síndrome constitucional LINFADENOPATÍAS Y ESPLENOMEGALIA Pancitopenia Anemia Infecciones Hemorragias Síndrome de la Vena Cava Superior Infiltración Meníngea Recidivas SNC** Testicular* Ocular y Pulmonar CD 19 + PAS + Tdt (frec + E)
")
24
1.a. L. Aguda Linfoblástica. SUBTIPOS
LAL COMÚN O Pre B - Más frecuente - Mejor pronóstico - Buen pronóstico: t(1;19) t(12;21) - Mal pronóstico: t(9;22) t(4;11) FORMA T NULAS FORMA B - Menos frecuente - Peor pronóstico - Traslocación (8;14) - Oncogén c-myc. Tdt (-)
 t(12;21) - Mal pronóstico: t(9;22) t(4;11) FORMA T. NULAS. FORMA B. - Menos frecuente. - Peor pronóstico. - Traslocación (8;14) - Oncogén c-myc. Tdt (-)")
25
1.a. LLA. PRONÓSTICO Y TTO Aspergilosis invasiva
FACTOR FAVORABLE DESFAVORABLE Edad Niños 1-9 años Adultos16-35 años Niños <1 y >10 años Adultos > 35 años Infiltración SNC No Si Leucocitos < 20000/mm3 > 50000/mm3 Citogenética Hiperploidía > 50 t (12;21) Hipodiploidía t(9,22)-Cr Philadelphia t(4,11) Blastos en mo a las 2 semanas del tto < 5% > 20% Aspergilosis invasiva Neumonía Pneumocysti carinii INDUCCIÓN PDN + VCR + ASP CONSOLIDACIÓN/MANTENIMIENTO MTX + 6-MP SNC MTX intratecal
 Hipodiploidía. t(9,22)-Cr Philadelphia t(4,11) Blastos en mo a las 2 semanas del tto. < 5% > 20% Aspergilosis invasiva. Neumonía Pneumocysti carinii. INDUCCIÓN. PDN + VCR + ASP. CONSOLIDACIÓN/MANTENIMIENTO. MTX + 6-MP. SNC. MTX intratecal.")
26
1.b. Leucemia Aguda No Linfoblástica
FACT. MAL PRONÓSTICO Cromosoma Ph; t(9,11) Enf. Preleucémica (SMD) > 60 años Formas 2ª a QT Aberraciones moleculares Leucostasis Manifestaciones Isquémicas BASTONES DE AUER
 Enf. Preleucémica (SMD) > 60 años. Formas 2ª a QT. Aberraciones moleculares. Leucostasis. Manifestaciones. Isquémicas. BASTONES. DE. AUER.")
27
ARA-C 1.b. Leucemia Aguda No Linfoblástica
Criterios de Bajo Riesgo t(8;21), inv 16 INDUCCIÓN ARA-C + antraciclina +/- etopósido MANTENIMIENTO ARA-C (citarabina) Riesgo elevado fact. mal pronóstico TMO M3 ATRA
, inv 16. INDUCCIÓN ARA-C + antraciclina +/- etopósido. MANTENIMIENTO ARA-C (citarabina) Riesgo elevado fact. mal pronóstico. TMO. M3. ATRA.")
28
Infiltración piel, encías y SNC
SUBTIPO FAB % C. Auer Peroxidasa Esterasa PAS Citometría de Flujo Citogenética Clínica M0; no diferenciada 2-3 CD13, 33 M1; sin maduración 20 + CD13, 33, 34, HLA-DR M2; con maduración 25 +++ CD13, 15, 33, 34, HLA-DR t(8;21) gen AML01-ETO M3; promielocítica 10 CD13, 15, 33 t(15;17) gen PML/RAR CID M4; mielomonocítica CD11b, 13, 14, 15, 33, HLA-DR M4Eo; inv 16 Infiltración piel, encías y SNC M5; monocítica t(11;23) M6; eritroleucémica 5 CD33, HLA-DR Fibrosis mo M7; megacariocítica ++ CD33, 41 L1; blastos pequeños 75 LAL preB CD10 (CALLA+) TDT+ t(9;22) y otras Hepatoesplenomegali. Infiltración de SNC y testículos. Adenopatías L2; blastos grandes LAL T FA+, TDT+ CALLA- L3; vacuolado TDT´- t(8;14)
 gen AML01-ETO. M3; promielocítica. 10. CD13, 15, 33. t(15;17) gen PML/RAR. CID. M4; mielomonocítica. CD11b, 13, 14, 15, 33, HLA-DR. M4Eo; inv 16. Infiltración piel, encías y SNC. M5; monocítica. t(11;23) M6; eritroleucémica. 5. CD33, HLA-DR. Fibrosis mo. M7; megacariocítica. ++ CD33, 41. L1; blastos pequeños. 75. LAL preB CD10 (CALLA+) TDT+ t(9;22) y otras. Hepatoesplenomegali. Infiltración de SNC y testículos. Adenopatías. L2; blastos grandes. LAL T FA+, TDT+ CALLA- L3; vacuolado. TDT´- t(8;14)")
29
Derivan de la MÉDULA ÓSEA
Clínica: EXPRESIÓN EN SANGRE PERIFÉRICA Diagnóstico: BIOPSIA MÉDULA ÓSEA LEUCEMIA AGUDA SÍNDROMES MIELOPROLIFERATIVOS CRÓNICOS GAMMAPATÍAS MONOCLONALES LLA (*) LANL/LMA LMC Policitemia Vera Trombocitemia Esencial Mielofibrosis idiopática Mieloma Múltiple Macroglobulinemia de Waldestron MGUS Enf. de las cadenas pesadas
 LANL/LMA. LMC. Policitemia Vera. Trombocitemia Esencial. Mielofibrosis idiopática. Mieloma Múltiple. Macroglobulinemia de Waldestron. MGUS. Enf. de las cadenas pesadas.")
30
2. Síndromes Mieloproliferativos Crónicos
PV MF TE LMC HEMATÍES N N o LEUCOCITOS o PLAQUETAS FOSFATASA ALCALINA LEUCOCITARIA o N FIBROSIS M.O. +++ ESPLEMOMEGALIA + CROMOSOMA PH. - MUTACIÓN JAK2 Hiperuricemia LDH Vit.B12

31
2.a. Leucemia Mieloide Crónica
LEUCOCITOSIS MADURA ( ) 30% crisis blástica IMATINIB 400 mg Dasatinib // Nilotinib CROMOSOMA PHILADELPHIA (Cr 22) REORDENAMIENTO bcr-abl
 30% crisis blástica. IMATINIB 400 mg. Dasatinib // Nilotinib. CROMOSOMA PHILADELPHIA (Cr 22) REORDENAMIENTO bcr-abl.")
32
2.b. Policitemia Vera EPO N o EPO SECUNDARIA PRIMARIA POLIGLOBULIA
TUMORES HIPOXEMIA Afinidad Hb-O2 PRIMARIA EPO N o POLICITEMIA VERA

33
Dos primeros A y dos parámetros B
2.b. Policitemia Vera. DIAGNÓSTICO CRITERIOS CARACTERÍSTICAS A1 Masa eritrocitaria 25% por encima de los valores medios de referencia A2 Exclusión de causas de policitemia secundaria A3 Esplenomegalia palpable A4 Positividad marcadores clonales (anormalidades citogenéticas) B1 Trombocitosis /mm3 B2 Leucocitosis neutrofílica (10.000/mm3) en ausencia de fiebre o infección B3 Esplenomegalia evidenciada por ecografía o radioisótopos B4 Bajos niveles séricos de eritropoyetina A1 + A2 + A3 o A4 ------ Dos primeros A y dos parámetros B
 B1. Trombocitosis /mm3. B2. Leucocitosis neutrofílica (10.000/mm3) en ausencia de fiebre o infección. B3. Esplenomegalia evidenciada por ecografía o radioisótopos. B4. Bajos niveles séricos de eritropoyetina. A1 + A2 + A3 o A Dos primeros A y dos parámetros B.")
34
2.b. Policitemia Vera. TRATAMIENTO
TROMBOSIS † SANGRÍAS AAS ANAGRELIDE ANTI PROLIFERATIVO HIDROXIUREA ÁC. ÚRICO PRURITO ERITROMELALGIA ALOPURINOL CIPROHEPTADINA AAS

35
(Metaplasia Mieloide Bazo) Leucoeritroblastosis
PLAQUETAS > HEMORRAGIA disfuncionants DIAGNÓSTICO POR EXCLUSIÓN HIDROXIUREA ANAGRELIDE 2.c. Trombocitemia Esencial 2.d. Mielofibrosis Idiopática HEMATOPOYESIS EXTRAMEDULAR (Metaplasia Mieloide Bazo) ANEMIA MIELOPTÍSICA Dacriocitos Leucoeritroblastosis BIOPSIA DIAGNÓSTICA (PAMO blanca) DD con MIELOFIBROSIS SECUNDARIAS HIDROXIUREA TPH
 ANEMIA MIELOPTÍSICA. Dacriocitos. Leucoeritroblastosis. BIOPSIA DIAGNÓSTICA. (PAMO blanca) DD con. MIELOFIBROSIS. SECUNDARIAS. HIDROXIUREA. TPH.")
36
Derivan de la MÉDULA ÓSEA
Clínica: EXPRESIÓN EN SANGRE PERIFÉRICA Diagnóstico: BIOPSIA MÉDULA ÓSEA LEUCEMIA AGUDA SÍNDROMES MIELOPROLIFERATIVOS CRÓNICOS GAMMAPATÍAS MONOCLONALES LLA (*) LANL/LMA LMC Policitemia Vera Trombocitemia Esencial Mielofibrosis idiopática Mieloma Múltiple Macroglobulinemia de Waldestron MGUS Enf. de las cadenas pesadas
 LANL/LMA. LMC. Policitemia Vera. Trombocitemia Esencial. Mielofibrosis idiopática. Mieloma Múltiple. Macroglobulinemia de Waldestron. MGUS. Enf. de las cadenas pesadas.")
37
3. Gammapatía Monoclonal
PARAPROTEINEMIA ORINA Ig MÉDULA ÓSEA MIELOMA +++ G > A > cadenas ligeras Cél. Plasmáticas WALDESTRÖM No o poca M Cél. Linfoplasmocitarias MGUS Cualquiera

38
3.a. Mieloma Múltiple. CLÍNICA
ASINTOMÁTICA DOLOR ÓSEO** INFECCIONES AFECTACIÓN RENAL VSG Anemia Paraproteinemia INSUFICIENCIA MO HIPERCALCEMIA HIPERVISCOSIDAD Astenia Anorexia Náuseas y vómitos Poliuria Polidipsia Estreñimiento Confusión

39
3.a. Mieloma Múltiple. DIAGNÓSTICO
CRITERIOS MAYORES Plasmocitoma en biopsia tisular MO: plasmáticas > 30% Pico monoclonal sérico > 3’5 g/dl si es IgG, 2 si es IgA o una proteinuria de cadenas ligeras > 1 g/día MENORES Celularidad en MO 10 – 30 % Pico monoclonal < al criterio mayor Lesiones osteolíticas radiológicas De inmunoglobulinas normales

40
ESTADIOS SEGÚN DURIE-SALMON Clasificación basada en
3.a. Mieloma Múltiple. CLASIFICACIÓN ESTADIOS SEGÚN DURIE-SALMON ESTADIO CONDICIONES MASA TUMORAL I Todas las enumeradas: Hemoglobina > 10g/dl Calcemia < 12 mg/dl Rx normal o con lesión única Paraproteína poco elevada: IgG < 5 g/dl IgA < 3 g/dl Cadenas ligeras en orina < 4 g/dl Baja II No cumple ni I ni III Intermedia III Uno o más de: Hemoglobina < 8’5 g/dl Calcemia > 12 mg/dl Lesiones osteolíticas intensas Paraproteína muy elevada: IgG > 7 g/dl IgA > 5 g/dl Cadenas ligeras en orina > 12 g/dl Alta A: Creatinina < 2 mg/100 ml B: Creatinina > 2 mg/100 ml Clasificación basada en 2 microglobulina I 2microglobulina < 3’5 + albúmina > 3’5 II ni I ni II III 2microglobulina > 5’5

41
Asintomático NO se trata, al igual que la LLC
3.a. Mieloma Múltiple. TRATAMIENTO INDUCCIÓN A LA REMISIÓN BORTEZOMIB + DEXAMETASONA INTERFERON DEXAMETASONA + LENALIDOMIDA BORTEZOMIB + LENALIDOMIDA MANTENIMIENTO LENALIDOMIDA LENALIDOMIDA + BORTEZOMIB Asintomático NO se trata, al igual que la LLC

42
Venas Arrosariadas “en Salchicha”
3.b. Macroglobulinemia Waldeström Monoclonal B IgM HIPERVISCOSIDAD Fondo de Ojo Venas Arrosariadas “en Salchicha” Lesiones Líticas Hipercalcemia Trisomía 12 3.c. MGUS Ancianos. 10% > 75 años Componente M 5 -6 % de cél plasmáticas en m o NO clínica Transformación MM 10-20% 3.d. Enfermedad de las Cadenas Pesadas

43
Diferencias entre MM y GMSi
MIELOMA MÚLTIPLE GMSi FRECUENCIA + ++++ SÍNTOMAS, SIGNOS Y COMPLICACIONES Dolor óseo, lesiones óseas, compresión radicular o medular, polineuropatía, infecciones IR, insuficiencia de la mo, hipercalcemia, hiperviscosidad (alteraciones neurológicas, visuales, hemorrágicas, ICC…) Asintomático por definición. No hay anemia, ni insuficiencia renal, ni hipercalcemia ni lesión ósea. PROTEINURIA DE BENCE-JONES ++ Menos frecuente e intensa ÍNDICE DE TIMIDINA TRITIADA > 1 % < 1 % CELULARIDAD PLASMÁTICA EN MO > 10% (criterio menor) > 30% (criterio mayor) < 10% EPIDEMIOLOGÍA Más frecuente en edad media o avanzada 1% de la población > 50 años 10% de la población > 75 años COMPONENTE M Pico monoclonal sérico > 3’5 g/dl si es IgG, 2 d/dl si es IgA, o proteinuria de cadena ligera > 1 g/día (criterio mayor) Pico monoclonal sérico inferior al criterio mayor (criterio menor) Pico monoclonal sérico < 3g/dl Proteinura de Bence-Jones en orina generalmente negativa PRONÓSTICO Malo Bueno TRATAMIENTO Si es asintomático no requiere tto Si tiene manifestaciones clínica QT No requiere tratamiento 25% evoluciona a MM
 Asintomático por definición. No hay anemia, ni insuficiencia renal, ni hipercalcemia ni lesión ósea. PROTEINURIA DE BENCE-JONES. ++ Menos frecuente e intensa. ÍNDICE DE TIMIDINA TRITIADA. > 1 % < 1 % CELULARIDAD PLASMÁTICA EN MO. > 10% (criterio menor) > 30% (criterio mayor) < 10% EPIDEMIOLOGÍA. Más frecuente en edad media o avanzada. 1% de la población > 50 años. 10% de la población > 75 años. COMPONENTE M. Pico monoclonal sérico > 3’5 g/dl si es IgG, 2 d/dl si es IgA, o proteinuria de cadena ligera > 1 g/día (criterio mayor) Pico monoclonal sérico inferior al criterio mayor (criterio menor) Pico monoclonal sérico < 3g/dl. Proteinura de Bence-Jones en orina generalmente negativa. PRONÓSTICO. Malo. Bueno. TRATAMIENTO. Si es asintomático no requiere tto. Si tiene manifestaciones clínica QT. No requiere tratamiento. 25% evoluciona a MM.")
44
TPH (Trasplante de Progenitores Hematopoyéticos)
EICH Aguda TMO Alogénico Piel: exantema eritrodermia Hígado: ibtericia obstructiva Intestino: naúseas, diarrea Crónica Liquen plano-esclerodermiforme Fallo hepatocelular Malabsorción Bronquiolitis obliterante

45
LOS GRANDES LOGROS SE CONSIGUEN PASO A PASO MUCHAS GRACIAS Y SUERTE!!

Presentaciones similares
















, bcl2 s Ig, CD19,CD20,CD21 SOBREVIDA 7-9 AÑOS TRASFORMACION.>")


