Descargar la presentación
La descarga está en progreso. Por favor, espere

1
CLASIFICACIÓN A- HIPERFENILALANINEMIAS - FENILCETONURIAS - TIROSINEMIAS - ALBINISMO B- SISTEMAS DE TRANSPORTE ALTERADOS - CISTINURIA - HARTNUP - FANCONI C- ACIDEMIAS ORGANICAS - ALCAPTONURIA - DE CADENA RAMIFICADA - LÁCTICO ACIDEMIAS

2
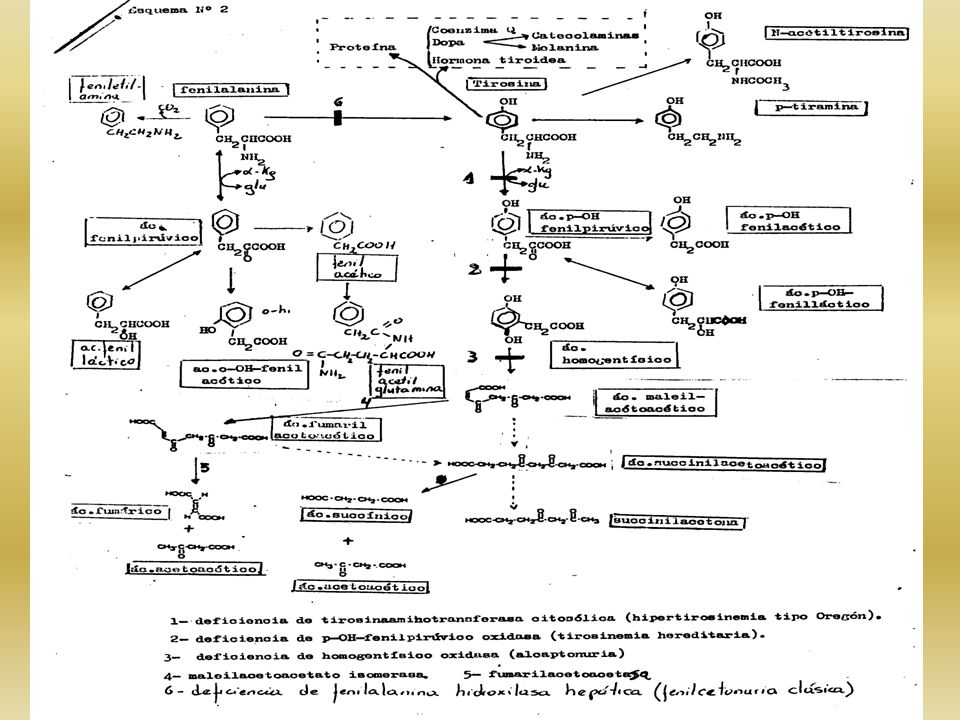
DESÓRDENES DEL METABOLISMO DE TIROSINA
PRECURSOR DE: HORMONAS TIROIDEAS – CATECOLAMINAS – MELANINA CLASIFICACIÓN 1- TIROSINEMIA TIPO OREGON DEFIC. DE TIROSINA AMINOTRANSFERASA 2- TIROSINEMIA HEREDITARIA NEONATAL HEPATORENAL CONGÉNITA DEFIC. DE P-OH-FENILPIRÚVICO OXIDASA 3- TIROSINEMIA TRANSITORIA DEL RN

4
1- TIROSINEMIA TIPO OREGON 2- TIROSINEMIA HEPATORENAL
DEFIC. ENZIMÁTICA ABSOLUTA DE TAT CITOSÓLICA COMPENSACIÓN POR AE MITOCONDRIAL ORINA: P- OH FP, POR TRANSAMINACIÓN EN LA MITOCONDRIA HIPERTIROSINURIA – HIPERTIROSINEMIA RETARDO MENTAL - MICROCEFALIA 2- TIROSINEMIA HEPATORENAL DEFIC. ABS. DE P-OH FPOXIDASA AGUDA O NEONATAL: - PRIMEROS 6 MESES – SIN TRATAMIENTO MUERE - VÓMITOS – DIARREA – HEMORRAGIAS – HEPATOMEGALIA - HIPOGLUCEMIA – RAQUITISMO FORMA CRÓNICA: - SEVERA CIRROSIS Y NEFROPATÍA - HIPERPLASIA DE ISLOTES DE LANGERHANS HIPOGLUCEMIA

6
ASPECTOS BIOQUÍMICOS ORINA: TIROSINURIA, P-OH-FENILACÉTICO Y LÁCTICO ALTERADA REABSORCIÓN TUBULAR PROTEINURIA - GLUCOSURIA – HIPERFOSFATURIA – AAC. GENERALIZADA DAÑO HEPÁTICO: HIPERMETIONINEMIA PLASMA DEL RN: ALFA-FETOPROTEÍNA (DAÑO HEPATOCELULAR) Y P-OH-FP DIAGNÓSTICO ACTIV. DE P-OH-FP OXIDASA EN BIOPSIA HEPÁTICA DETECCIÓN DE TIR Y SUS METAB. EN ORINA LA EXCRECIÓN DE d - ALA PBG EN ORINA PARA DESCARTAR PAI (FUERTES DOLORES ABDOMINALES) DIAGNÓSTICO PRENATAL: DETER. DE FAH EN CULTIVO DE CÉLULAS DE LÍQUIDO ANMIÓTICO
 Y P-OH-FP. DIAGNÓSTICO. ACTIV. DE P-OH-FP OXIDASA EN BIOPSIA HEPÁTICA. DETECCIÓN DE TIR Y SUS METAB. EN ORINA. LA EXCRECIÓN DE d - ALA. PBG EN ORINA PARA DESCARTAR PAI (FUERTES DOLORES ABDOMINALES) DIAGNÓSTICO PRENATAL: DETER. DE FAH EN CULTIVO DE CÉLULAS DE LÍQUIDO ANMIÓTICO.")
7
3- TIROSINEMIA TRANSITORIA
FRECUENTE – INMADUREZ GESTACIONAL PREMATUROS: % TIR RN A TÉRMINO: % TIR LETARGIA Y MOVIMIENTOS ERRÁTICOS TRATAMIENTO: REDUCIR LA INGESTA PROTEICA A 2-3 G/KG/DÍA CIRROSIS HEPÁTICA CON DISFUNCIÓN RENAL: TIROSINEMIA HEREDITARIA (P-OH FP OXIDASA) FRUCTOSURIA (F-1-P ALDOLASA) GALACTOSEMIA (GAL-1-P-URIDIL TRANSFERASA) RAQUITISMO POR DEFECTO TUBULAR
 FRUCTOSURIA (F-1-P ALDOLASA) GALACTOSEMIA (GAL-1-P-URIDIL TRANSFERASA) RAQUITISMO POR DEFECTO TUBULAR.")
8
ALCAPTONURIA 1859: SE OBSERVA QUE LA ORINA DE LOS ENFERMOS SE PONIA OSCURA CUANDO SE DEJABA AL AIRE SE LLAMA A LA SUSTANCIA : ALCAPTÓN (AVIDEZ POR EL OXÍGENO EN MEDIO ALCALINO) Y FINALMENTE SE IDENTIFICA COMO 2,5-DI-OH FENILACÉTICO ÁC. HOMOGENTÍSICO LA ENZIMA DEFICIENTE ES : HOMOGENTÍSICO OXIDASA
 Y FINALMENTE SE IDENTIFICA COMO 2,5-DI-OH FENILACÉTICO ÁC. HOMOGENTÍSICO. LA ENZIMA DEFICIENTE ES : HOMOGENTÍSICO OXIDASA.")
9
CUADRO CLÍNICO: EN LA INFANCIA LOS PACIENTES SON ASINTOMÁTICOS EL PIGMENTO OCRONÓTICO SE VA DEPOSITANDO EN FORMA INTRA Y EXTRACELULAR APARECE ÁCIDO HOMOGENTÍSICO EN ORINA NO AUMENTA EN SANGRE PORQUE EL RIÑÓN LO EXCRETA MECANISMO DE DEFENSA REQUIEREN AÑOS HASTA LA APARICIÓN DE OCRONOSISPIGMENTACIÓN DE CARTÍLAGOS, ESCLERÓTICA, CONJUNTIVA, CÓRNEA

10
EL PIGMENTO ES UN POLÍMERO DEL ÁCIDO HOMGENTÍSICO
EN TEJIDO CONECTIVO PRODUCE ALTERACIONES ARTRITIS OCRONÓTICA CARTÍLAGOS AFECTADOS: TRAQUEALES, LARÍNGEOS, COSTALES, TENDONES, LIGAMENTOS. EL PIGMENTO APARECE EN LA ZONA GENITAL Y AXILAR, ES MARRÓN Y MANCHA LA ROPA. NO SE ELIMINAN OTROS AAC POR ORINA

12
DIAGNÓSTICO 1- APARICIÓN DE COLOR OSCURO EN ORINA EN PRESENCIA DE ÁLCALIS, SE OSCURECE PRIMERO LA SUPERFICIE 2- CON RVO. DE BENEDICT EN ½ OH, VIRA NARANJA MARRÓN 3- REDUCCIÓN DE MOLIBDATO 4- REACCIÓN CON Cl3Fe ROJO PÚRPURA 5- COLORACIÓN NEGRA DE LA ORINA AL ADICIONAR NITRATO DE PLATA 6- CONFIRMACIÓN: CROMATOGRAFÍA EN CAPA FINA

13
PRONÓSTICO: LOS PACIENTES DESARROLLAN OCRONOSIS SUFREN INVALIDEZ ENTRE LOS 50 – 70 AÑOS CAMBIOS DEGENERATIVOS EN LAS ARTICULACIONES TRATAMIENTO: - NO HAY UNA VEZ ESTABLECIDA LA OCRONISIS LA TERAPIA SE INCLINA A CORREGIR LA ARTRITIS SE RESTRINGE FEN Y TIR DE LA DIETA Y SE ADM VIT C ÁCIDO HOMOGENTÍSICO INHIBE LA ENZIMA LISIL-HIDROXILASA, QUE PARTICIPA EN EL METABOLISMO DEL COLÁGENO EN PRESENCIA DE ASCORBATO NO HAY INHIBICIÓN

14
ALBINISMO GRUPO DE ENFERMEDADES QUE AFECTAN AL SISTEMA PIGMENTARIO DE MELANINA CARACTERÍSTICAS CLÍNICAS: FOTOFOBIA – NISTAGMUS – AGUDEZA VISUAL – AUSENCIA O MARCADA DE MELANINA EN PIEL, CABELLO Y OJOS TRASTORNOS EN LA COAGULACIÓN Y SISTEMA DE MACRÓFAGOS PROCESOS INFECCIOSOS, ALTERADA FUNCIÓN DE LISOSOMAS Y FAGOLISOSOMAS SÍNTESIS DE MELANINA: - MELANINA MELANOSOMAS MELANOCITOS ASOCIADOS CON UN POOL DE 36 QUERATINOCITOS EPIDERMIS

15
LA SÍNTESIS DE MELANINA COMIENZA A PARTIR DE TIR Y TIROSINASA EN EL PREMELANOSOMA
MELANOSOMA SOLO CONTIENE MELANINA EUMELANOSOMAS COLOR MARRÓN Y NEGRO FEOMELANOSOMAS COLOR ROJO O AMARILLO TIROSINASA GLICOPROTEÍNA Cu DEPENDIENTE – DOPA : COFACTOR DE LA REACCIÓN DE MELANOGÉNESIS LAS ALTERACIONES EN LA SÍNTESIS DE MELANINA PUEDEN SER: A NIVEL DE TIROSINASA POST- TIROSINASA

17
COLOR DE LA PIEL ORIGEN RACIAL DEL INDIVIDUO TIPOS DE ALBINISMO:
EL PIGMENTO DE LA PIEL HUMANA TIENE 2 COMPONENTES: 1- COLOR CONSTITUTIVO CANTIDAD DE PIGMENTO GENERADO POR UN PROGRAMA GENÉTICO EN AUSENCIA DE LUZ UV INTERVIENEN 3 O 4 PARES DE GENES (BLANCO AL NEGRO) 2- COLOR FACULTATIVO INDUCIBLE POR LA LUZ UV, HORMONAS Y ALGUNAS ENFERMEDADES – ES REVERSIBLE, SIN INFLUENCIA GENÉTICA COLOR DE LA PIEL ORIGEN RACIAL DEL INDIVIDUO TIPOS DE ALBINISMO: OCULO- CUTÁNEO 10 FORMAS DESCRIPTAS PIEL- OJOS- CABELLOS OCULAR 4 FORMAS DESCRIPTAS – AFECTA SOLAMENTE OJOS
 2- COLOR FACULTATIVO INDUCIBLE POR LA LUZ UV, HORMONAS Y ALGUNAS ENFERMEDADES – ES REVERSIBLE, SIN INFLUENCIA GENÉTICA. COLOR DE LA PIEL ORIGEN RACIAL DEL INDIVIDUO. TIPOS DE ALBINISMO: OCULO- CUTÁNEO 10 FORMAS DESCRIPTAS PIEL- OJOS- CABELLOS. OCULAR 4 FORMAS DESCRIPTAS – AFECTA SOLAMENTE OJOS.")
18
ALBINISMO OCULO-CUTÁNEO
MANIFESTACIONES CLÍNICAS: AGUDEZA VISUAL – FOTOFOBIA –NISTAGMUS 1- TIROSINASA (-) 2- TIROSINASA (+) 3- SÍNDROME DE HERMANSKY- PUDLAK 4- SÍNDROME DE CHEDIAK-HIGASHI 5- SÍNDROME DE CROSS 6- ALBINISMO OCULO-CUTÁNEO MARRÓN 7- ALBINISMO OCULO-CUTÁNEO BERMEJO 8- ALBINISMO OCULO-CUTÁNEO AD 9- ALBINISMO NEGRO SENSONEURAL 10- ALBINISMO MUTANTE AMARILLO
 2- TIROSINASA (+) 3- SÍNDROME DE HERMANSKY- PUDLAK. 4- SÍNDROME DE CHEDIAK-HIGASHI. 5- SÍNDROME DE CROSS. 6- ALBINISMO OCULO-CUTÁNEO MARRÓN. 7- ALBINISMO OCULO-CUTÁNEO BERMEJO. 8- ALBINISMO OCULO-CUTÁNEO AD. 9- ALBINISMO NEGRO SENSONEURAL. 10- ALBINISMO MUTANTE AMARILLO.")
19
TIROSINASA (-) FORMA CLÁSICA – AUSENCIA COMPLETA DE PIGMENTO MELANOCITOS CON MELANOSOMAS NO PIGMENTADOS NO HAY SÍNTESIS DE TIROSINASA LOS NIVELES SÉRICOS DE TIR, CU, Y HME SON NORMALES CABELLO BLANCO NIEVE, PIEL ROSADA, OJOS ROSADOS LOS BULBOS DE LOS PELOS, COMO FUENTE DE ENZIMA, INCUBADOS CON L-TIR, NO FORMAN PIGMENTO

20
SÍNDROME DE HERMANSKY-PUDLAK
TIROSINASA (+) PIEL Y OJOS AFECTADOS EXISTE ALGO DE PIGMENTO VISIBLE: PELO BLANCO O AMARILLO TOSTADO BULBO PILOSO INCUBADO CON TIR DEPOSITA MELANINA HAY SÍNTESIS DEL PIGMENTO PERO NO SE PUEDE LIBERAR DEFECTO POST-TIROSINASA DETERMINAR LA ACTIVIDAD ENZIMÁTICA SÍNDROME DE HERMANSKY-PUDLAK TIR (+), CON MÍNIMA FORMACIÓN DE PIGMENTO HEMORRAGIAS GINGIVALES PLAQUETAS SIN TROMBOXANO A2 AGREGACIÓN PLAQUETARIA
 PIEL Y OJOS AFECTADOS. EXISTE ALGO DE PIGMENTO VISIBLE: PELO BLANCO O AMARILLO TOSTADO. BULBO PILOSO INCUBADO CON TIR DEPOSITA MELANINA. HAY SÍNTESIS DEL PIGMENTO PERO NO SE PUEDE LIBERAR. DEFECTO POST-TIROSINASA. DETERMINAR LA ACTIVIDAD ENZIMÁTICA. SÍNDROME DE HERMANSKY-PUDLAK. TIR (+), CON MÍNIMA FORMACIÓN DE PIGMENTO. HEMORRAGIAS GINGIVALES. PLAQUETAS SIN TROMBOXANO A2 AGREGACIÓN PLAQUETARIA.")
21
SÍNDROME DE CHEDIAK- HIGASHI
TIR (+), DEFECTO 1° DESCONOCIDO – FATAL EN LA NIÑEZ MACROMELANOSOMAS – MENOS PIGMENTO CABELLO GRIS-METÁLICO INFECCIONES Y NEUROPATÍAS PERIFÉRICAS LEUCOCITOS CON ENZIMAS LISOSOMALES LA CÉLULA NO PUEDE ELIMINAR LOS MATERIALES FAGOCITADOS ALBINISMO OCULAR CLÁSICO – LIGADO A X PIGMENTO DEL IRIS – NISTAGMUS – FOTOFOBIA - MACROMELANOSOMAS
, DEFECTO 1° DESCONOCIDO – FATAL EN LA NIÑEZ. MACROMELANOSOMAS – MENOS PIGMENTO. CABELLO GRIS-METÁLICO. INFECCIONES Y NEUROPATÍAS PERIFÉRICAS. LEUCOCITOS CON ENZIMAS LISOSOMALES LA CÉLULA NO PUEDE ELIMINAR LOS MATERIALES FAGOCITADOS. ALBINISMO OCULAR. CLÁSICO – LIGADO A X. PIGMENTO DEL IRIS – NISTAGMUS – FOTOFOBIA - MACROMELANOSOMAS.")
22
AMINOACIDURIAS POR ALTERACIÓN DEL TRANSPORTE A TRAVÉS DE MEMBRANA
AMINOACIDURIAS RENALES SE HAN ESTABLECIDO 5 TIPOS DE SISTEMAS DE TRANSPORTE DE AMINOÁCIDOS ESPECÍFICOS DE GRUPO, A NIVEL DE RIÑÓN: 1- AAC. DICARBOXÍLICOS 2- AAC. DIBÁSICOS 3- AAC. ALIFÁTICOS Y AROMÁTICOS NEUTROS 4- IMINOÁCIDOS Y GLICINA 5- b- AMINOÁCIDOS ALGUNOS DE ESTOS MECANISMOS TB OPERAN EN INTESTINO

23
CISTINURIA AR ALTERADO EL TRANSPORTE DE AAC. A NIVEL DEL EPITELIO INTESTINAL Y DEL TÚBULO RENAL SE EXCRETAN POR ORINA CIS – ORN – LIS – ARG COMPARTEN EL MISMO SISTEMA DE TRANSPORTE LOS NIVELES EN PLASMA SON NORMALES O BAJOS NO HAY DEPÓSITO DE CIS EN TEJIDOS PÉRDIDA DE LA CAPACIDAD REABSORTIVA DEL TÚBULO RENAL

24
EXPERIMENTOS IN VITRO DEMOSTRARON LA DIVERSIDAD DE PROTEÍNAS CARRIERS PARA “C- O- L – A” EN RIÑÓN
TRABAJANDO CON VESÍCULAS DE MEMBRANAS SE OBS EN EL BORDE EN CEPILLO SISTEMAS DE TRANSPORTE DE AAC Na+ DPTES EN LA MEMBRANA BASOLATERAL EL TRANSPORTE ES POR DIFUSIÓN FACILITADA

25
SEGÚN EL SISTEMA DE TRANSPORTE ALTERADO, SON LAS DISTINTAS ANORMALIDADES:
TIPO I NO HAY TRANSPORTE DE O – L – A TIPO II NO SE ABSORBE NINGUNO DE LOS 4 AAC TIPO III NO SE ABSORBE CIS EN INTESTINO LOS AAC QUE NO SE ABSORBEN SON UTILIZADOS POR LA FLORA INTESTINAL L- O –A SON DESCARBOXILADOS POR LAS BACTERIAS Y PRODUCEN: DIAMINAS CADAVERINA Y PUTRECINA DE LA DEGRADACIÓN DE: L PIPERIDINA A PIRROLIDINA LA ABSORCIÓN DE L-CISTEÍNA ES NORMAL DISTINTO MECANISMO DE TRANSPORTE

26
PEQUEÑOS OLIGOPÉPTIDOS Y DIPÉPTIDOS SE ABSORBEN Y TRANSPORTAN NORMALMENTE EN ENFERMOS CISTINÚRICOS USAN DISTINTOS SISTEMAS DE TRANSPORTE ESTOS SUPLEN LA CARENCIA NUTRICIONAL OCASIONADA POR LA FALTA DE ABSORCIÓN DE AAC Y PROVEEN AAC ESENCIALES

27
CUADRO CLÍNICO: SI BIEN LOS 4 AAC SE EXCRETAN POR ORINA EN ELEVADA PROPORCIÓN EN PACIENTES HOMOCIGOTAS CISTINA ES RESPONSABLE DEL CUADRO CLÍNICO ES EL MENOS SOLUBLE PRECIPITA FORMANDO CÁLCULOS URETRALES Y RENALES OBSTRUCCIONES, INFECCIONES E INSUFICIENCIA RENAL ASOCIACIÓN CON HIPERTENSIÓN CUADRO MAS SEVERO EN EL HOMBRE ANATOMÍA DEL TRACTO URINARIO SI BIEN LA ENFERMEDAD SE MANIFIESTA EN EL 1° AÑO DE VIDA HACE PICO EN LA 2° O 3° DÉCADA LOS CÁLCULOS SON RADIOOPACOS DEBIDO A LA DENSIDAD DE LOS GRUPOS SULFURO

28
SOLUBILIDAD DE CISTINA EN FUNCIÓN DEL PH DE LA ORINA
CISTINA ES INSOLUBLE A PH 3- 5 CON LA ALCALINIZACIÓN SE SOLUBILIZA Y APARECE EN GRANDES CANTIDADES EN ORINA DIAGNÓSTICO: PRIMERA ORINA DE LA MAÑANA MÁS CONCENTRADA 1- OBS DEL SEDIMENTO URINARIO CÁLCULOS, CRISTALES HEXAGONALES DE COLOR AMARILLO- MARRÓN 2- TEST DEL NITROPRUSIATO DE SODIO 3- CROMATOGRAFÍA EN CAPA FINA LAS PIEDRAS SE FORMAN CON EXCRESIÓN DE CIS MAYOR DE 300MG/G DE CREATININA EN ORINA NORMAL 75 A 125 MG/G DE CREATININA

29
TRATAMIENTO 1- RESTRICCION DIETARIA PARA DISMINUIR LA PRODUCCIÓN Y EXCRESIÓN DE ESTOS AAC 2- AUMENTAR LA SOLUBILIDAD DE CIS POR ALCALINIZACIÓN DE LA ORINA, ADMINISTRANDO BICARBONATO O CITRATO 3- AUMENTAR EL VOLUMEN DE ORINA, BEBIENDO HASTA 4 LTS. AGUA/DÍA PARA AUMENTAR LA DIURESIS 4- AUMENTAR LA EXCRESIÓN DE CIS POR EL USO DE D- PENICIL-AMINA (b,b – DIMETILCISTEÍNA) QUE FORMA UN DI SULFURO MEZCLA CISTEÍNA- PENICILAMINA, MAS SOLUBLE SE EXCRETA POR ORINA - ESTE TRATAMIENTO SI BIEN ES EFECTIVO, AL SER PROLONGADO CON EL TIEMPO PROVOCA PROTEINURIA Y SÍNDROME NEFRÓTICO
 QUE FORMA UN DI SULFURO MEZCLA CISTEÍNA- PENICILAMINA, MAS SOLUBLE SE EXCRETA POR ORINA. - ESTE TRATAMIENTO SI BIEN ES EFECTIVO, AL SER PROLONGADO CON EL TIEMPO PROVOCA PROTEINURIA Y SÍNDROME NEFRÓTICO.")
30
ALA, SER, TREO, LEU, VAL, ISOLEU, TRIP
DESÓRDEN DE HARTNUP ALTERADO SISTEMA DE TRANSPORTE PARA AAC NEUTROS, MONOAMINAS, MONOCARBOXÍLICOS EN INTESTINO Y TÚBULO RENAL AMINOACIDURIA MASIVA SE EXCRETAN 5 A 20 VECES MAS ALA, SER, TREO, LEU, VAL, ISOLEU, TRIP COMPARTEN EL MISMO SISTEMA DE REABSORCIÓN RENAL. A NIVEL INTESTINAL NO SON REABSORBIDOS ATAQUE BACTERIANO LUEGO DE UNA DIETA RICA EN TRIP SE EXCRETAN GRANDES CANTIDADES DE INDOLACÉTICO E INDICÁN ORINA Y HECES COLOR AMARILLENTAS TRIP ES PRECURSOR DE NIACINA SU PROVOCA ALTERACIONES DERMATOLÓGICAS Y SÍNTOMAS NEUROLÓGICOS DEL TIPO PELAGRA

31
SE SUGIERE QUE EL DEFECTO A NIVEL INTESTINAL Y RENAL CONDUCE A:
FORMACIÓN DE PRODUCTOS DE DESCOMPOSICIÓN TÓXICOS PARA EL SNC MENOR CANTIDAD DE NICOTINAMIDA PELAGRA MENOR DISPONIBILIDAD DE AAC ESENCIALES MALNUTRICIÓN MENOR DISPONIBILIDAD DE TRIP SUSTRATO PARA LA SÍNTESIS DE SEROTONINA

32
SÍNDROME DE FANCONI HIPERAMINOACIDURIA GENERALIZADA POR INHIBICIÓN DEL TRANSPORTE TUBULAR SE OBSERVA: - DISFUNCIÓN COMPLEJA DE LOS TÚBULOS PROXIMALES MAYOR CLEARENCE RENAL DE : FOSFATO, BICARBONATO, GLU, ÁCIDO ÚRICO - ALTERADA REABSORCIÓN DE Na+, Cl-, H2O Y CO3H ALTERACIÓN EQUILIBRIO ELECTROLÍTICO - DEFECTO METABÓLICO ÓSEO RAQUITISMO EN NIÑOS Y OSTEOMALACIA EN ADULTOS - HIPOCALCEMIA - HIPERPARATIROIDISMO ASOCIADOS TERAPIA: - ADM DE VIT D MEJORA EL CUADRO RENAL CORRIGE LA HIPOCALCEMIA

33
ALTERACIÓN EN EL METABOLISMO DE LOS AAC DE CADENA RAMIFICADA
LOS AAC NEUTROS RAMIFICADOS: LEU, ISOLEU Y VAL (ESENCIALES, ALIFÁTICOS), SON CATABOLIZADOS POR MECANISMOS ANÁLOGOS: 1- TRASAMINACIÓN 2- DESCARBOXILACIÓN OXIDATIVA 3- DESHIDROGENACIÓN HAY DEFICIENCIA DE LA DESHIDROGENASA (COMPLEJO ENZIMÁTICO) LOS ALFA-CETOÁCIDOS SE ACUMULAN EN SANGRE Y PASAN A ORINA OLOR CARACTERÍSTICO ENFERMEDAD CON OLOR A JARABE DE ARCE - EN SANGRE Y ORINA AUMENTA LA CONCENTRACIÓN DE LEU, ISOLEU Y VAL POR REVERSIBILIDAD DE LA TRASAMINASA , ENZIMA MUY ACTIVA EN TEJIDOS EXTRAHEPÁTICOS
, SON CATABOLIZADOS POR MECANISMOS ANÁLOGOS: 1- TRASAMINACIÓN. 2- DESCARBOXILACIÓN OXIDATIVA. 3- DESHIDROGENACIÓN. HAY DEFICIENCIA DE LA DESHIDROGENASA (COMPLEJO ENZIMÁTICO) LOS ALFA-CETOÁCIDOS SE ACUMULAN EN SANGRE Y PASAN A ORINA OLOR CARACTERÍSTICO ENFERMEDAD CON OLOR A JARABE DE ARCE. - EN SANGRE Y ORINA AUMENTA LA CONCENTRACIÓN DE LEU, ISOLEU Y VAL POR REVERSIBILIDAD DE LA TRASAMINASA , ENZIMA MUY ACTIVA EN TEJIDOS EXTRAHEPÁTICOS.")
34
CUADRO CLÍNICO HETEROGENEIDAD CLÍNICA 5 FENOTIPOS POR ALELOS MUTANTES QUE OCUPAN EL MISMO LOCUS -LA FORMA CLÁSICA SEVERA CETOACIDOSIS EN LOS PRIMEROS DÍAS DE NACIDO, CON SIGNOS NEUROLÓGICOS CONVULSIONES – RIGIDEZ – VÓMITOS – RESPIRACIÓN IRREGULAR EL ENFERMO NO TRATADO COMA MUERTE INCIDENCIA 1:120000 AR TRATAMIENTO: NO ES SENCILLO PORQUE INVOLUCRA 3 AAC ESENCIALES DIETAS RICAS EN ESTOS AAC + PROTEÍNAS NATURALES PARA PROMOVER EL DESARROLLO ADM TIAMINA QUE ESTABILIZA EL COMPLEJO ENZIMÁTICO RECORDAR: DEFICIENCIA DE TIAMINA BERIBERI DEFICIENCIA DE NIACINA PELAGRA DEFICIENCIA DE VIT C ESCORBUTO

Presentaciones similares





>")








. Forma primaria: herencia autosómica recesiva Alteración.>")







