Descargar la presentación
La descarga está en progreso. Por favor, espere

1
Ataxias heredodegenerativas.
Dr Alex Espinoza Giacomozzi. Neurología. Hospital DIPRECA. Reunión clínica.

2
Son un grupo de desórdenes caracterizado por incoordinación y pérdida del balance, a menudo acompañados de otra sintomatología neurológica. Son entidades usualmente progresivas, neurodegenerativas y llevan a la discapacidad y finalmente a la muerte.

3
Aproximación clínica. Confirmar si la ataxia está presente.
Descartar causas tratables. Obtener reporte confirmado. Es conocido el gen alterado. Hay clara historia familiar. Consejo genético y manejo sintomático. Buscar causas infrecuentes. Estudio genético.

4
Diagnóstico. Evaluar si el pcte presenta ataxia.
De ser así se deben descartar causas adquiridas. Si existe historia familiar, se debe estudiar patrón genético. Causas secundarias en general: Condición nutricional, estructural, endocrina, tóxicas, paraneoplásica e inflamatoria

5
¿Existe historia familiar?
Las ataxias secundarias tienen un desarrollo lento de síntomas (subagudo), cambios no específicos a la imaginología. No existe historia familiar: Paraneoplásica o inflamatoria. ¿Existe historia familiar? La respuesta es obvia cuando existe una clara historia familiar. Investigar a lo menos 3 generaciones: Forma de inicio de los síntomas. Edad de inicio. Consanguineidad, abortos espontáneos, origen étnico.
, cambios no específicos a la imaginología. No existe historia familiar: Paraneoplásica o inflamatoria. ¿Existe historia familiar La respuesta es obvia cuando existe una clara historia familiar. Investigar a lo menos 3 generaciones: Forma de inicio de los síntomas. Edad de inicio. Consanguineidad, abortos espontáneos, origen étnico.")
6
En términos generales una de las características genéticas de estas alteraciones es la anticipación genética. Las generaciones sucesivas son afectadas con más severidad. A edades más tempranas (expresividad). Aumenta la penetrancia (probabilidad de desarrollar la enfermedad teniendo la mutación). En lo molecular, se produce un mecanismo mutacional dinámico de polinucleótidos repetidos inestables.
. Aumenta la penetrancia (probabilidad de desarrollar la enfermedad teniendo la mutación). En lo molecular, se produce un mecanismo mutacional dinámico de polinucleótidos repetidos inestables.")
8
¿Se debe solicitar estudio genético a todos los pctes con Sd atáxico progresivo?
Opinión de expertos es controversial. Estudios han demostrado que entre un 5 a 15% de pctes con “ataxias adquiridas” tenían una hereditaria. Moseley ML et al, Neurology 51: 1666–1671, 1998. Soong BW et al, Arch Neurol 58: , 2001.

9
Causas de historia familiar negativa en desórdenes hereditarios.
Inadecuada obtención de historia familiar. Herencia ligada a X o recesiva. Penetrancia reducida. Anticipación genética. Variabilidad genética. Nueva mutación. Falsa paternidad. Muertes tempranas.

10
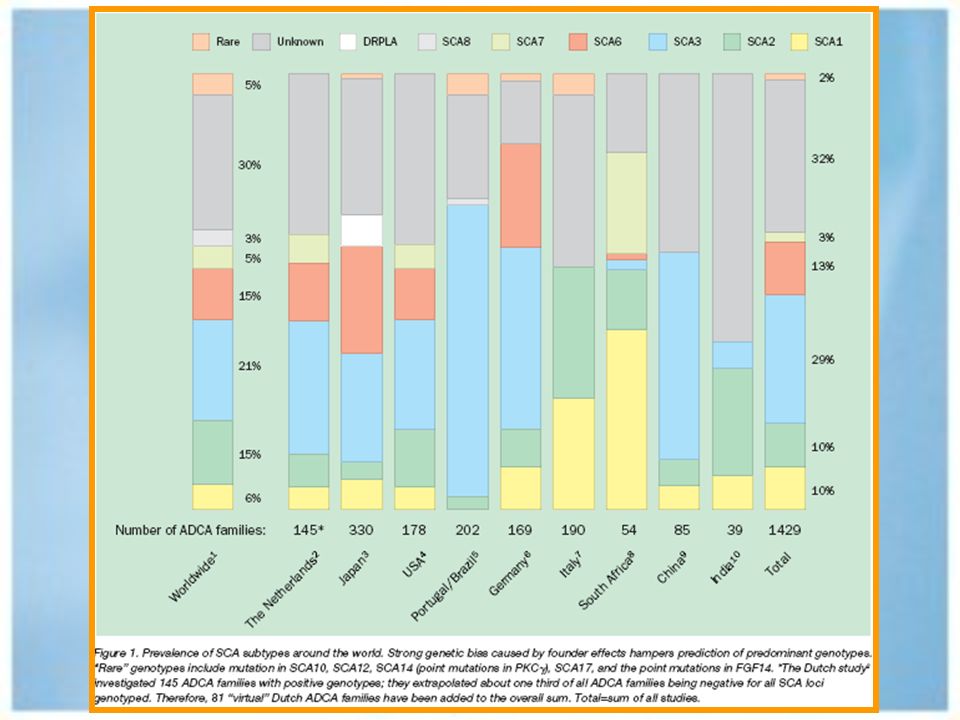
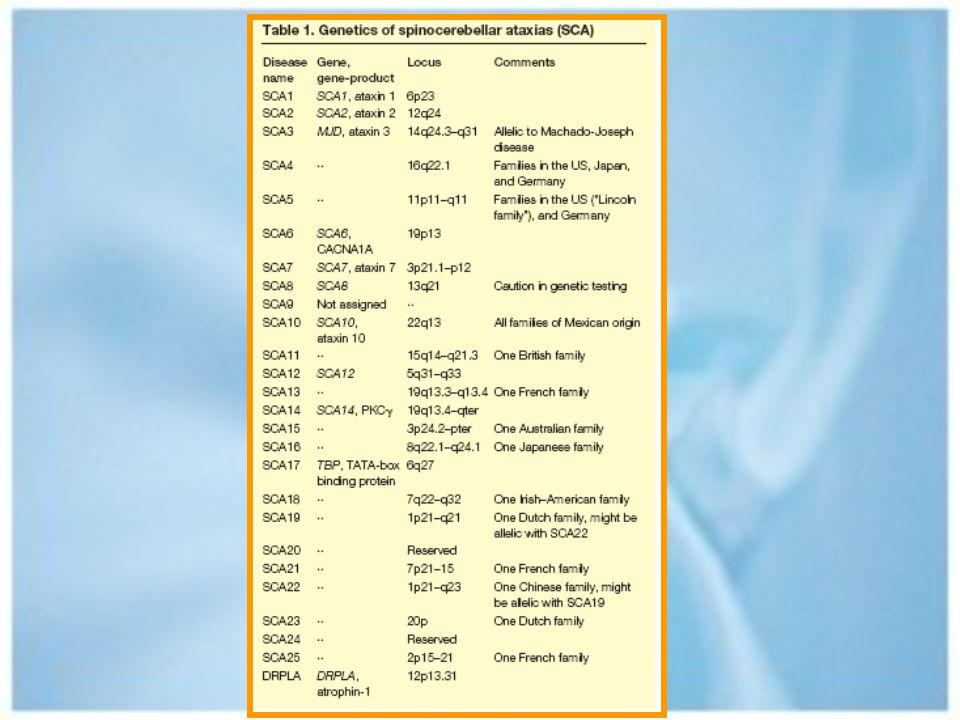
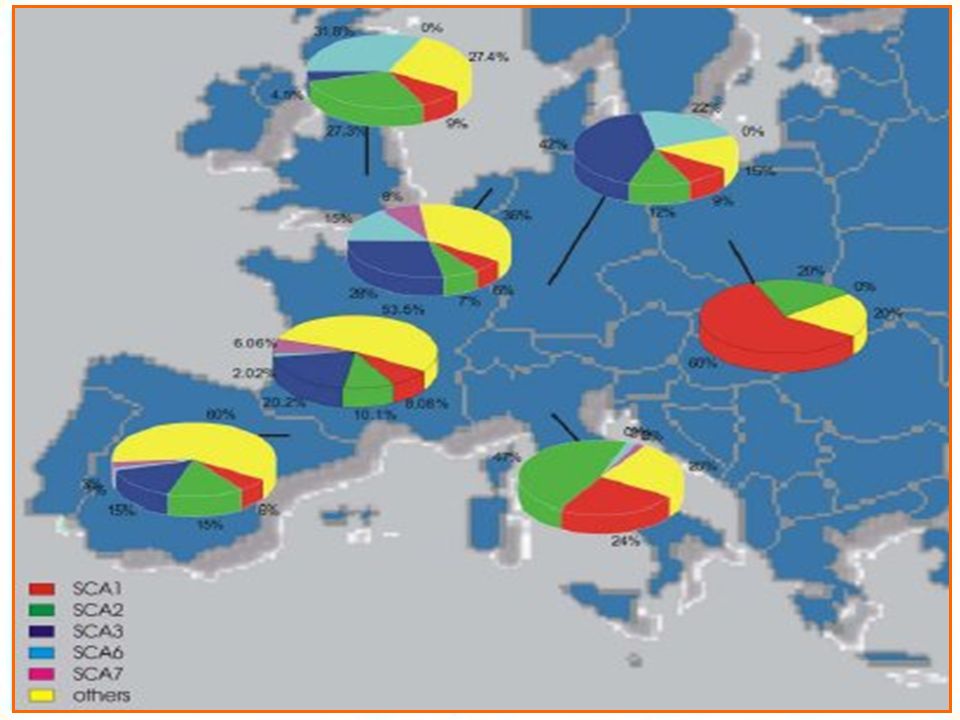
Ataxias hereditarias. Autosómicas dominante. Autosómicas recesivas.
Ligadas a X. Mitocondriales.

11
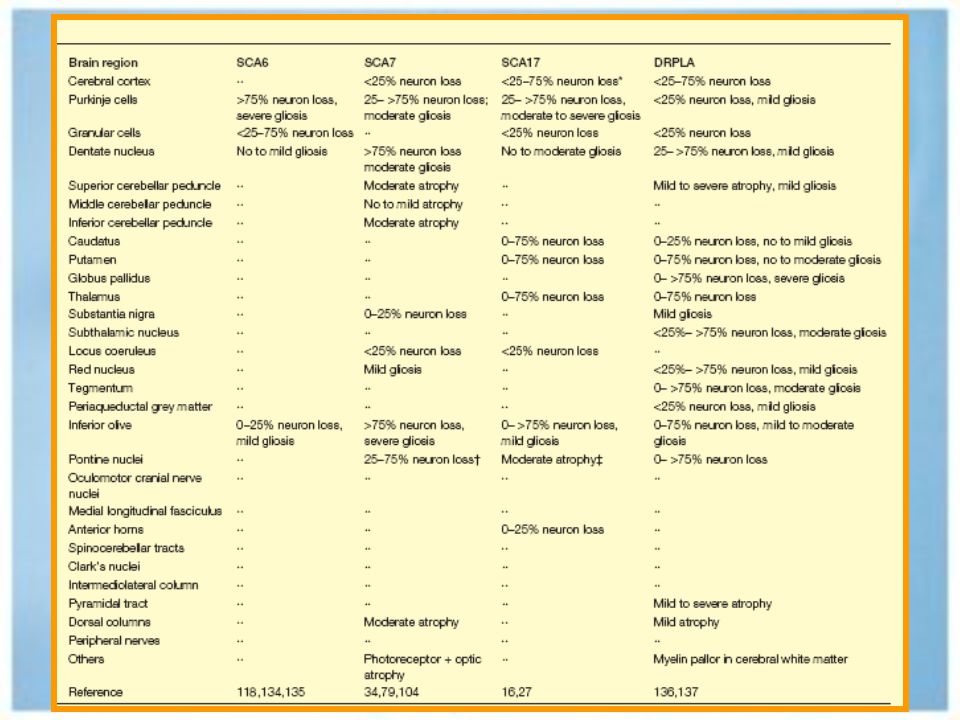
Sustrato anatomopatológico.
Atrofia olivopontocerebelosa Semiología cerebelosa y disartria. Luego la lesión afecta a neocorteza, ganglios de la base, sustancia nigra semiología extracerebelosa. Atrofia cerebelosa cortical SCA 5, 6, 10 y ataxia cerebelosa pura. Atrofia espinopontina SCA 3.

12
Atrofia dentatorubropalidolusyana sistema cerebeloso y palidofugales.
Degeneración espinocerebelosa SCA 4.

15
Ataxias autosómicas dominantes.
Ataxia espinocerebelosa tipo 1. Ataxia espinocerebelosa tipo 2. Ataxia espinocerebelosa tipo3/ Enf de Machado – Joseph. Ataxia espinocerebelosa 4 hasta 17. Atrofia Dentatorubropalidoluysiana. Ataxia episódica tipo 1 y tipo 2.

20
Ataxia espinocerebelosa tipo 1.
Más frecuente en 3 a 4ta década (aunque amplia). Clínica: Se inicia sólo por una leve pérdida del balance, progresando a ataxia de extremidades, disatria y sd psudobulbar. Nistagmus está presente precozmente, luego sácadas lentas y parálisis. Sd piramidal tb es precoz. La hiperreflexia puede ser reemplazada por hiporreflexia debido a neuropatía.
. Clínica: Se inicia sólo por una leve pérdida del balance, progresando a ataxia de extremidades, disatria y sd psudobulbar. Nistagmus está presente precozmente, luego sácadas lentas y parálisis. Sd piramidal tb es precoz. La hiperreflexia puede ser reemplazada por hiporreflexia debido a neuropatía.")
21
En lo extrapiramidal lo llamativo es el corea, que aparece en etapas tardías.
Compromiso cognitivo también es tardío. Causa de fallecimiento, complicación respiratoria por sd pseudobulbar. Sobrevida 10 a 15 años. En lo molecular esta enfermedad se produce por acumulación de tripletes CAG (39 – 81) en el gen SCA 1, produciéndose acumulación de glutamina en la proteína ataxina 1, de función desconocida.
 en el gen SCA 1, produciéndose acumulación de glutamina en la proteína ataxina 1, de función desconocida.")
22
Ataxia espinocerebelosa tipo 2.
Descrita en población Cubana, es la más frecuente en UK, Italia e India. Presenta overlap con la SCA tipo 1. Se presenta en la cuarta década. Clínica: La ataxia y disartria están siempre presentes. Lentitud de sácadas. Temblor, corea, distonía, hiporreflexia y demencia (tardíos). Ocurre una mutación del gen SCA 2 (12q), consistente en una expansión CAG (34 – 59), produciéndo la ataxina 2, presente en el citoplasma.
. Ocurre una mutación del gen SCA 2 (12q), consistente en una expansión CAG (34 – 59), produciéndo la ataxina 2, presente en el citoplasma.")
23
Ataxia espinocerebelosa tipo 3/ enfermedad de Machado – Joseph.
Descrita en portugueses. Clínica: Tipo I: sd espástico + bradiquinesia + distonía + leve ataxia. Tipo II: más común, ataxia + alteración de 1ra motoneurona. Tipo III: ataxia + neuropatía periférica. Tipo IV: parkinsonismo puro. La tipo I es más frecuente en jóvenes y la III en mayores.

24
La edad de inicio y la duración de la enfermedad es igual a SCA 1 y 2.
Existe gran variabilidad intrafamiliar. Causado por expansión CAG (54 – 86) en 14q, causando la proteína ataxina 3 cuya función normal es desconocida, pero se sabe que es predominantemente citoplasmática.
 en 14q, causando la proteína ataxina 3 cuya función normal es desconocida, pero se sabe que es predominantemente citoplasmática.")
25
Ataxia espinocerebelosa tipo 4.
Raro subtipo. Clínica: Ataxia + neuropatía axonal sensitiva. Ocasionalmente presente, signos piramidales. El gen responsable es desconocido, pero estaría localizado 16 q.

26
Ataxia espinocerebelosa tipo 5.
Llamada ataxia de Lincoln. Raro subtipo. Se iniciaría entre la 3 a 4 ta década. Clínica: Disfunción cerebelosa pura de lenta evolución. En etapas avanzadas sd pseudobulbar. El gen mutado estaría en 11q. Al igual que la tipo 4 aún no se conoce la expansión anómala.

27
Ataxia espinocerebelosa tipo 6.
Se debe a la mutación del gen de la CACNA1A (19p), subunidad 1 del canal de calcio voltaje dependiente. Migraña hemipléjica familiar y ataxia episódica tipo 2. Clínica: Al inicio, tinnitus, nauseas, vértigo, ataxia, disartria y alteraciones visuales. En forma episódica. Posteriormente se desarrolla una ataxia progresiva. Raro, oftalmoplegia, alteración sensitiva y extrapiramidal. A diferencia de otras SCA, su inicio es más tardío, curso más benigno.
, subunidad 1 del canal de calcio voltaje dependiente. Migraña hemipléjica familiar y ataxia episódica tipo 2. Clínica: Al inicio, tinnitus, nauseas, vértigo, ataxia, disartria y alteraciones visuales. En forma episódica. Posteriormente se desarrolla una ataxia progresiva. Raro, oftalmoplegia, alteración sensitiva y extrapiramidal. A diferencia de otras SCA, su inicio es más tardío, curso más benigno.")
28
Ataxia espinocerebelosa tipo 7.
Lo distintivo de este subtipo es la retinopatía progresiva irreversible que produce una ceguera bilateral. Se produce alteración de gen en 3p. Parte como una degeneración macular con un core central pigmentado, que luego afecta a la periferia. Puede presentarse discromatopsia para azul – amarillo. Falla visual es en el centro del campo visual. Clínica: Ataxia, disartria, alteración de la agudeza visual, signos de 1 ra neurona, apalestesia y alteración oculomotora.

29
Ataxia espinocerebelosa tipo 8.
Tiene mucha variabilidad de penetrancia. Se afecta gen del 13q. Clínica: Signos de 1ra motoneurona, apalestesia, ataxia y disartria. Se produce una repetición altamente inestable CTG, más grave cuando es transmitido por la madre. Afecta la transcripción del RNA no la formación de una proteína.

30
Ataxia espinocerebelosa tipo 10.
Pesquisado en población Mexicana. Clínica: Sd cerebeloso puro. Crisis convulsivas. Raro, sd piramidal, diskinesia ocular, alteraciones cognitivas y polineuropatía. Se ha descrito en algunas familias alteración cardíaca y hepática. La mutación es un pentanucleótido ATTCT en el 22q.

31
Ataxia espinocerebelosa tipo 11.
Clínica: Hiperreflexia + ataxia. Localizado en el 15q. El gen no ha sido identificado.

32
Ataxia espinocerebelosa tipo 12.
Inicio sobre los 40 años. Expansión CAG (55 a 78) en 5q. En un gen que codifica para una subunidad regulatoria de la fosfatasa 2A. Clínica: Temblor de extremidades. Disfunción cerebelar. Demencia en etapas avanzadas.
 en 5q. En un gen que codifica para una subunidad regulatoria de la fosfatasa 2A. Clínica: Temblor de extremidades. Disfunción cerebelar. Demencia en etapas avanzadas.")
33
Ataxia espinocerebelosa tipo 13 – 16.
Descrita en una familia francesa. Clínica: Inicio en la niñez - adolescencia. Ataxia. Retardo mental moderado. Defecto en el cromosoma 19q. SCA 14 es una ataxia pura progresiva en adultos mayores. SCA 15 (familia australiana) ataxia pura leve. SCA 16 (familia japonesa) temblor cabeza y ataxia progresiva.
 ataxia pura leve. SCA 16 (familia japonesa) temblor cabeza y ataxia progresiva.")
34
Atrofia Dentatorubropalidolusiana.
Se caracteriza por una amplia variedad fenotípica. Mas frecuente en la población japonesa. Clínica: Ataxia. Mioclonus. Coreoatetosis. Alteraciones psiquiátricas. Epilepsia. Demencia. La edad de inicio se relaciona con la clínica.

35
Los pctes que inician el cuadro antes de los 20 años siempre tienen crisis epilépticas, epilepsia mioclónica progresiva. Cuando el cuadro se inicia después de los 40 años, las crisis son raras, lo que más se asocia es la coreoatetosis, con demencia en etapas avanzadas. Presenta anticipación genética ocurre principalmente en la transmisión paterna. Se afecta el gen de la ataxina 1 en 12p (expansión CAG). Se observan inclusiones intracitoplasmáticas y nucleares.
. Se observan inclusiones intracitoplasmáticas y nucleares.")
37
Ataxia episódica tipo 1. Aparece durante la niñez o adolescencia.
Clínicas: Ataques breves de ataxia que duran de segundo a minutos generalmente precipitados por el ejercicio. Sin vértigo. Mioclonía continua interictal. Se afecta un canal potasio voltaje dependiente KCNA 1. Tb puede producir miokimia + epilepsia parcial. Pueden presentar mejoría con acetazolamina, fenitoína y carbamacepina.

38
Ataxia episódica tipo 2. Se debe a mutación CACNA1A, que codifica para la subunidad 1 del canal de calcio voltaje dependiente. Clínica: Disartria. Vértigo. Nauseas. Oscilopsia. Diplopia. Anormalidades interictales, sd cerebeloso, progresivo. Factores precipitantes, café, stress, OH. Manejo con acetazolamida.

41
Ataxias autosómicas recesivas.
Ataxia de Friedreich. Ataxia telangectasia. Ataxia asociada a déficit de vitamina E. Ataxia con apraxia oculomotora. Ataxia espástica autosómica recesiva de Charlevoix – Saguenay. Ataxia espinocerebelosa con neuropatía axonal.

42
Ataxia de Friedreich. Causa más común de ataxia hereditaria.
1: – en caucásicos. Clínica: Ataxia progresiva que inicia antes de 25 años. Disartria dentro de 5 años. Arreflexia en EEII, neuropatía axonal sensitiva. Sd piramidal. Anestesia profunda. Miocardiopatía (10 – 15%), diabetes (20%), escoliosis, intolerancia a la glucosa (50%).
, diabetes (20%), escoliosis, intolerancia a la glucosa (50%).")
43
Se produce una expansión GAA en un intrón del gen FRDA, que codifica para una proteína mitocondrial frataxina produciendo: Alteración del metabolismo de metales. Defecto en la fosforilación oxidativa. Aumenta stress oxidativo. Tratamiento: Antioxidante coenzima Q10 (400 mg/ día) y vitamina E (2100 mg/d).
 y vitamina E (2100 mg/d).")
46
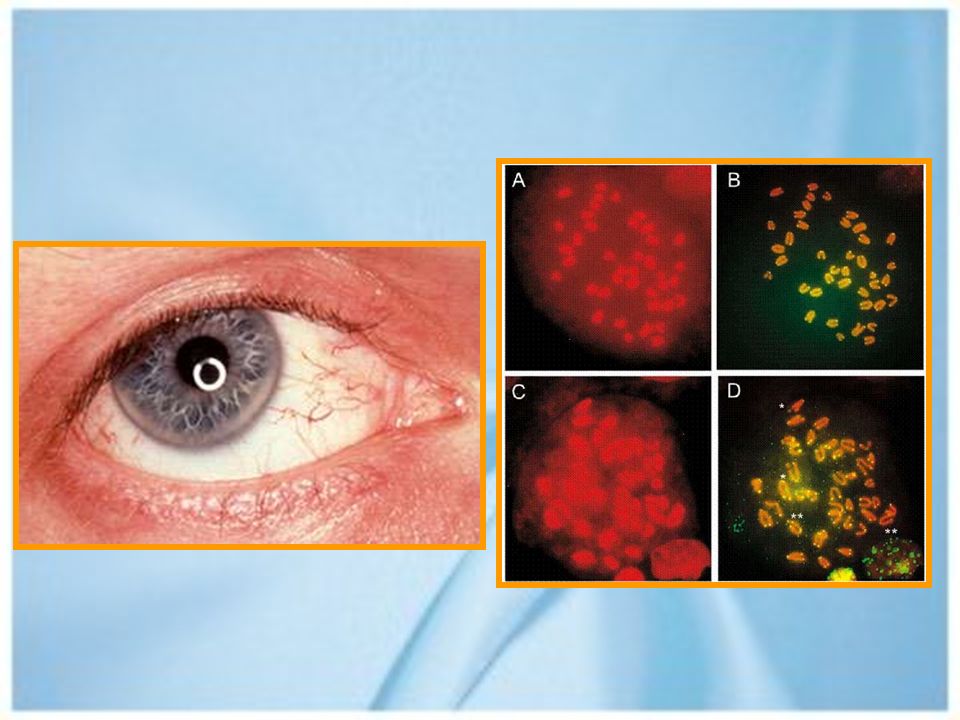
Ataxia telangectasia. Segunda causa de ataxia infantil. Clínica:
Inicio precoz. Telangectasia oculocutánea (95%). Deficiencias inmunológicas. Predisposición a malignidad (40%). Ataxia progresiva, disartria, hipotonía facial. Apraxia ocular y fijación inestable. Distonía, coreoatetosis. Tardío: signos de 2da motoneurona.
. Deficiencias inmunológicas. Predisposición a malignidad (40%). Ataxia progresiva, disartria, hipotonía facial. Apraxia ocular y fijación inestable. Distonía, coreoatetosis. Tardío: signos de 2da motoneurona.")
47
Las alteraciones cognitivas si ocurren son leves y en forma muy tardía.
Existe elevación de la alfafetoproteína. Ocurre un defecto del gen ATM, que normalmente produce una proteín kinasa implicada en la regulación del ciclo celular, apoptosis y reparación del DNA.

52
Ataxia asociada a deficiencia de vitamina E.
1.- Por mutación de la proteína de transporte del alfa tocoferol. Clínica es similar a la ataxia de Friedreich, pero con menor miocardiopatía. 2.- Por Abetalipoproteinemia. Se muta una proteína microsomal transportadora de triglicéridos.

53
Ataxia con apraxia oculomotora.
Muy similar a la ataxia telangectasia, pero sin las alteraciones extraneurales. Clínica: Ataxia progresiva en la niñez. Apraxia oculomotora. Arreflexia con neuropatía sensitiva axonal. Corea. Hipoalbuminemia e hipercolesterolemia. Mutación del cromosoma 9p, que codifica para la apratoxina (repara DNA).
.")
54
Ataxia espástica autosómica recesiva de Charlevoix – Saguenay.
Rara. Clínica: Ataxia precoz + espasticidad + amiotroifia distal. Alteración de la mirada de seguimiento horizontal.

55
Ataxia espinocerebelosa con neuropatía axonal.
La última caracterizada. Se afectan genes encargados de codificar para proteínas de reparación del DNA (fosfodiesterasa tirosyl – DNA 1). Clínica: Ataxia de inicio precoz. Neuropatía axonal. Hipercolesterolemia, hipoalbuminemia.
. Clínica: Ataxia de inicio precoz. Neuropatía axonal. Hipercolesterolemia, hipoalbuminemia.")
56
Ataxias ligadas a X. Anemia sideroblástica + ataxia mutación en el gen ABC7, que codifica para una transportador de metales mitocondrial. Sd X frágil. Por repetición CGG en el cromosoma X. Presentan temblor, ataxia y parkinsonismo leve. Poseen cuerpos de inclusión ubiquinina +.

57
Ataxias mitocondriales.
Mutación en el gen para la ATPasa 6. Debilidad neurogénica. Ataxia. Retinitis pigmentaria. Sd de Leigh. Fibras rasgadas están ausentes en la biopsia muscular. Pueden no tener elevación de lactato o piruvato.

58
Clasificación antigua (harding).
ADCA I: SCA 1, 2, 3, 4, 8, 13 y 14. ADCA II: SCA 7. ADCA III: SCA 5, 6, 11, 12 y 16.

60
Tratamiento sintomático.
El manejo de la ataxia raramente responde a algún tipo de tto, debido a la coordinación está regulada por una serie de mecanismos complejos. Existen estudios que reportan beneficios: Botez MI, et al: treatment of Friedreich ´s ataxia with amantadine. Neurology 39: , 1989. Takei et al: tandospirone en enf de Machado – Joseph. Psychiatry Clin N 56: 181 – 185, 2002. Síntomas extrapiramidales responden bien a agonistas dopaminérgicos, anticolinérgicos y botox. Gwinn – Hardy et al. Neurology 55: 800 – 805, 2000. Schos et al. Arch Neurol 57: 1495 – 1500, 2000.

61
Manejo de disfunción urinaria reponde a:
Oxibutirina. Tolterodina. Trastornos del sueño responden a tratamiento habitual. Dentro de las alteraciones psiquiátricas la depresión es común, menos común es la psicosis y la ansiedad. Importantísimo manejo kinésico y de terapia ocupacional.

Presentaciones similares























, aunque sus síntomas.>")


 Novedades>")





