Descargar la presentación
La descarga está en progreso. Por favor, espere

1
Síndrome QT largo

2
SQTL: Historia 1957: 1er reporte SQTL 1963-1964: Síndrome Romano-Ward
: 25 casos de SQTL reportados 1971: 1er Tto SQTL (Estelectomía izquierda) 1979: Comenzó el Registro de SQTL : + 10 genes responsables identificados Jervell and Lange-Nielsen provided the first documented description of this syndrome in They described a family with 4 deaf children with QT prolongation and syncope. 3 of the children died suddenly. The parents, and 2 other children had normal ECGs and normal hearing. This report was critical to the development of current knowledge regarding LQTS. Romano in 1963, and Ward in 1964 separately reported patients who were not deaf, but had an almost identical cardiac disorder. Later called the “Romano-Ward syndrome,” this disorder was realized to be much more common than the “Jervell-Lange-Nielsen syndrome.” The genetic transmission was thought to be autosomal dominant. Between 1958 and 1970, there are only 25 cases of LQTS reported. In 1971, Moss and McDonald performed the first successful therapy for LQTS in a patient who had not responded to any other antiarrhythmic therapy: left cardiac sympathetic denervation to shorten the QT interval. In 1979,Drs. Moss, Schwartz, and Crampton started the International Registry for LQTS, with headquarters in Rochester, NY. Between 1991 and 2001, a total of 6 LQTS genes were identified, 3 of which were reported between March 1995 and January 1996.
 1979: Comenzó el Registro de SQTL : + 10 genes responsables identificados. Jervell and Lange-Nielsen provided the first documented description of this syndrome in They described a family with 4 deaf children with QT prolongation and syncope. 3 of the children died suddenly. The parents, and 2 other children had normal ECGs and normal hearing. This report was critical to the development of current knowledge regarding LQTS. Romano in 1963, and Ward in 1964 separately reported patients who were not deaf, but had an almost identical cardiac disorder. Later called the Romano-Ward syndrome, this disorder was realized to be much more common than the Jervell-Lange-Nielsen syndrome. The genetic transmission was thought to be autosomal dominant. Between 1958 and 1970, there are only 25 cases of LQTS reported. In 1971, Moss and McDonald performed the first successful therapy for LQTS in a patient who had not responded to any other antiarrhythmic therapy: left cardiac sympathetic denervation to shorten the QT interval. In 1979,Drs. Moss, Schwartz, and Crampton started the International Registry for LQTS, with headquarters in Rochester, NY. Between 1991 and 2001, a total of 6 LQTS genes were identified, 3 of which were reported between March 1995 and January")
3
SQTL Congénito Múltiples mutaciones Hasta 10 tipos genéticos: LQT1-10 Herencia AD ó AR Terapia específica según genotipo. Desencadenadas por descarga adrenérgica Adquirido Múltiples causas: Metabólicas Farmacológicas Estructurales Se corrige tratando la causa

4
LQTS Congénito: Fenotipia - Romano-Ward (RW)
- Jervell-Lange-Nielsen (JLN) - Esporádico
 - Esporádico.")
5
QT largo Congénito Sme. RW
Autosómica dominante Audición normal Más frecuente de los QTL hereditarios

6
QT Largo Congénito Sme de JLN
Autosómico recesivo Asocia sordera neurosensorial 1.6 a 6 casos/millón de habitantes

7
QTL Congénito Formas esporádicas
10 al 15 % de los casos Audición normal Carácter hereditario no identificado Pentrancia incompleta???

10
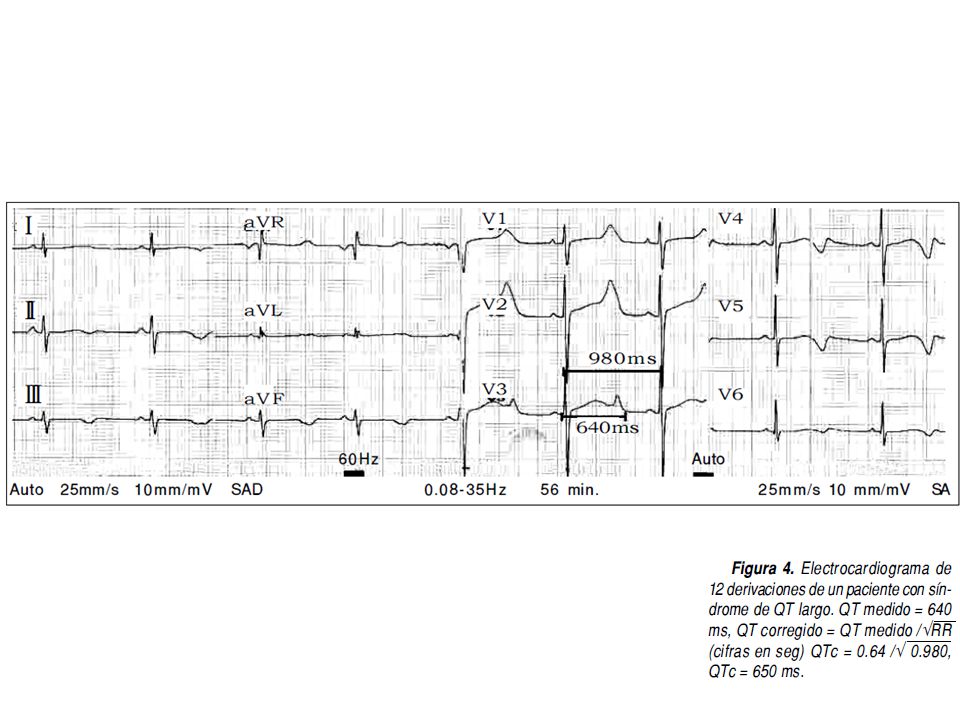
El síndrome de QT largo (SQTL) se caracteriza por una grave alteración en la repolarización ventricular traducida en el electrocardiograma (ECG) por un alargamiento en el intervalo QT. que predispone a arritmias ventriculares malignas —torsade de pointes— y muerte súbita.

11
CANALOPATÍAS QUE GENERAN SINDROME QT LARGO

13
3 2 1

14
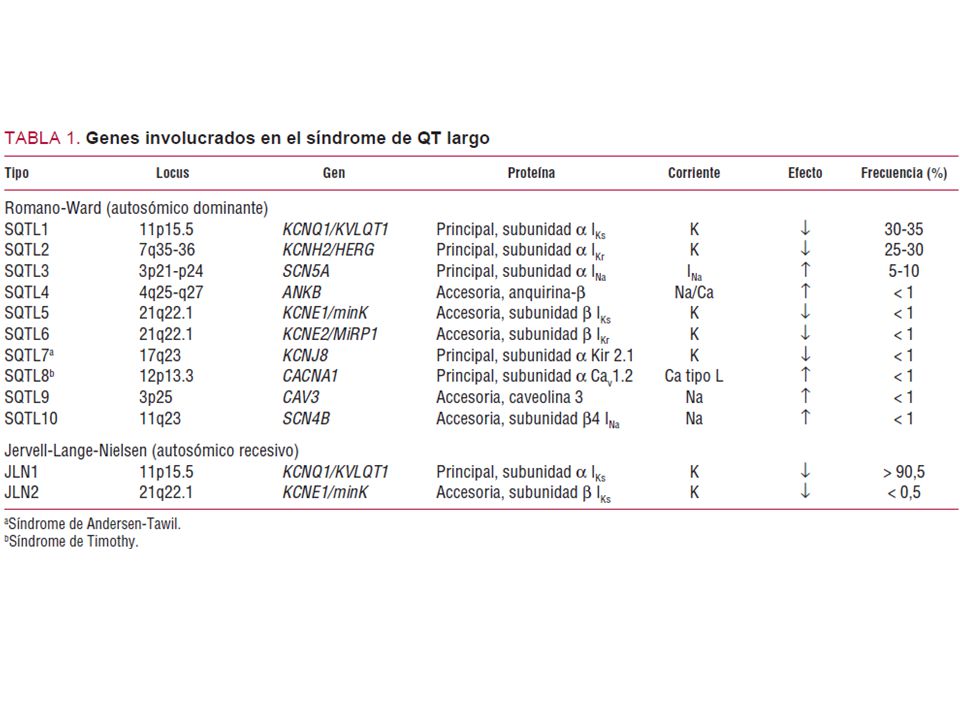
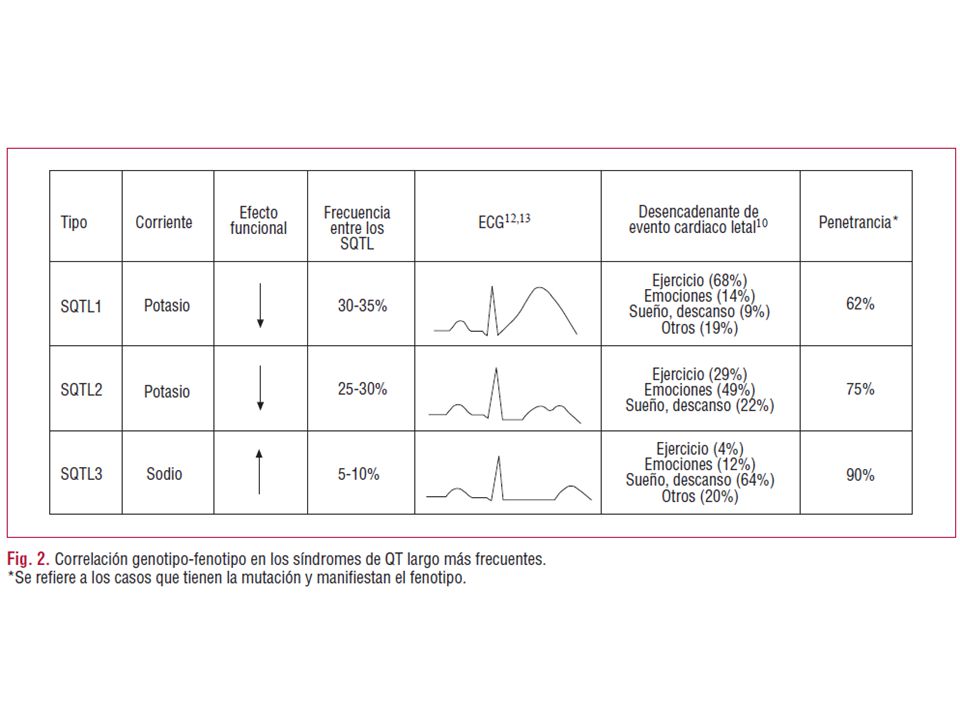
SQTL1 Onda T de base ancha larga (broad-based prolonged T waves). Dependencia moderada de la frecuencia cardíaca del intervalo QT. Afecta el brazo corto del cromosoma 11. Mutación: 11p15.5. Canal afectado en el potencial de acción: IKS delayed rectifier potassium current. Es la única variante con un porcentaje elevado de eventos durante los ejercicios físicos o la natación. SQTL2 Onda T con muescas y de voltaje reducida. Intervalo QT con dependencia moderada de la frecuencia cardíaca. kcnh2 en la mutación l413p el559h. SQTL3 Intervalo QT prolongado como consecuencia del aumento en la duración del segmento ST. Aparición tardía de la onda T. Dependencia significativa de la frecuencia cardíaca. Gen afetado: SCN5A; mutación p21-24 en el cromosoma 3. Fase del potencial de acción: fase 2, plateau o dome por entrada persistente de sodio.

15
Representación esquemática del complejo macromolecular.
Los canales iónicos son proteínas transmembranales (α) reguladas por diversas proteínas, una de ellas es la llamada subunidad β.
 reguladas por diversas proteínas, una de ellas es la llamada subunidad β.")
19
SQTL: Aspectos clínicos
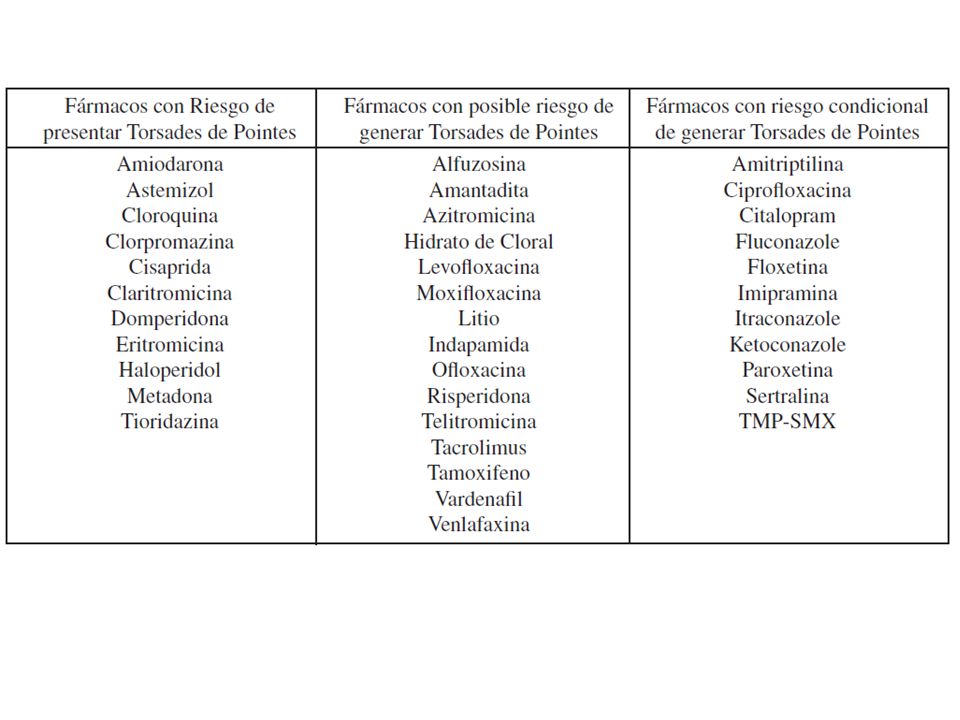
Signos y Síntomas: Síncope Convulsiones MS Palpitaciones o dolor precordial Symptoms include: The presence of palpitations or children may describe as “chest pain” Recurrent syncope Seizures SCD can occur if the symptoms are ignored ECG signs include: QTc abnormal or borderline Evidence of Torsade de pointes is also common Patients at higher risk are those with deafness, female, syncope, and documented torsades or VF.

20
Aortic Stenosis, HCM, Myxoma
Syncope Slow Onset Abrupt Onset Abrupt Onset Slow Offset Abrupt Offset Slow Offset Hyperventilation Seizure disorder Hypoglycemia Obstructive Arrhythmic Vascular If all other diagnoses have been ruled out, it may be helpful to evaluate the presentation of the syncopal episode, if this information is available. 1. Slow onset and slow offset When syncope comes on slowly, and fades away slowly, causes are usually related to an episode of hypoglycemia or hyperventilation. 2. Abrupt onset and abrupt offset The sudden onset and sudden termination of a syncopal episode usually indicates a cardiac cause. There are 3 major categories of cardiac causes: Obstructive origin: Could be related to aortic stenosis, hypertrophic cardiomyopathy, or myxoma Arrhythmic origin: Could be either a brady or tachyarrhythmia Vascular origin: Most common causes are orthostatic hyoptension or vasovagal attack 3. Abrupt onset and slow offset Episodes of syncope that start suddenly and then slowly dissipate are most often related to a seizure disorder. ECGs are recommended in almost all patients because the results can lead to decisions about immediate management of underlying disease, or can help identify further testing. Vasovagal, Orthostatic Hypertension Aortic Stenosis, HCM, Myxoma Brady Tachy

22
Bradicardia Sinusal Disfunción del nodo Sinusal:
SQTL1, SQTL3 (mas frecuente) SQTL4 > DISFUNCION SINUSAL
 SQTL4 > DISFUNCION SINUSAL.")
23
Alternancia Eléctrica de la onda T

24
Bloqueo AV 2x1

25
Taquicardia Helicoidal

27
Tratamiento Betabloqueadores Bloqueadores de canales de Sodio
Potasio suplementario y fármacos que facilitan su disponibilidad Marcapaso

Presentaciones similares