Descargar la presentación
La descarga está en progreso. Por favor, espere

1
GENERALIDADES EN DEMENCIAS
Dra.Yazmìn Mora Cambronero FISIOPATOLOGIA UNIBE

2
Bases Bioquímicas de las Enfermedades Neurodegenerativas

3
Proteostasis EQUILIBRIO
Proceso que mantiene el equilibrio entre la producción y la destrucción o reciclado de las proteínas corporales

4
AGREGACIÓN DE PROTEINAS
Desequilibrio AGREGACIÓN DE PROTEINAS

5
Enfermedades Neurodegenerativas
Comparten una característica común: El depósito o agregación de proteínas anormales extra o intracelularmente.

6
MECANISMOS DE DEFENSA CELULAR
Un fallo en la estructura de los polipéptidos representa un problema serio para la viabilidad y función celular Las neuronas son vulnerables al efecto tóxico de estas proteínas MUTANTES

7
MECANISMOS DE DEFENSA CELULAR
La célula se protege contra las proteínas mal ensambladas ¿ CÓMO ? Con varios tipos de mecanismos de defensa celular…

8
CHAPERONAS La primera línea de defensa contra las proteínas mal ensambladas. Promueven el correcto plegamiento de las proteínas conforme salen de los ribosomas. Las proteínas chaperonas son un conjunto de proteínas presentes en todas las células, muchas de las cuales son proteínas de choque térmico, cuya función es la de ayudar al plegamiento de otras proteínas recién formadas en la síntesis de proteínas. Estas chaperonas no forman parte de la estructura primaria de la proteína funcional, sino que sólo se unen a ella para ayudar en su plegamiento, ensamblaje y transporte celular a otra parte de la célula donde la proteína realiza su función. Los cambios de conformación tridimensional de las proteínas pueden estar afectados por un conjunto de varias chaperonas que trabajan coordinadas, dependiendo de su propia estructura y de la disponibilidad de las chaperonas.

9
Sistema Ubiquitina-Proteasoma
Degrada proteínas que no son plegadas o ensambladas correctamente Degrada proteínas dañadas o envejecidas” (vida media corta)
")
10
Proteasomas Se encarga de degradar proteínas nucleares y citosólicas mal ensambladas o mal plegadas. Las proteínas van marcadas por ubiquitina y luego el proteasoma las degrada. Las proteínas deben de estar desenrolladas antes de entrar al proteasoma.

11
SISTEMA UBIQUITINA-PROTEASOMA

12
UBIQUITINA - PROTEASOMA
Las enfermedades neurodegenerativas son llamadas proteinopatías porque su origen ocurre en la acumulación de alguna proteína y su eliminación depende del sistema de desintegración proteica: UBIQUITINA - PROTEASOMA

13
Fallo en el sistema Si no se detecta la proteína anormal
Factor inicial o acelerante de la enfermedad neurodegenerativa Complejos proteicos grandes no pueden entrar al sistema proteasoma para ser desintegrado Proteasoma no rompe enlaces poliglutamina de estas enfermedades

14
Enfermedades asociadas con fallo en el sistema
Acúmulos proteicos en Enfermedad de Alzheimer : Depósitos de Beta Amiloide Ovillos neurofibrilares (hiperfosforilación de proteína tau) Acúmulos proteicos en Enfermedad de Huntington: Lenta en producir acumulación tóxica
 Acúmulos proteicos en Enfermedad de Huntington: Lenta en producir acumulación tóxica.")
15
Sistema Lisosomal Macroautofágico
Elimina organelas enteras y complejos multiproteicos Este mecanismo incluye formación de estructura de doble membrana AUTOFAGOSOMA El autofagosoma se fusiona con lisosoma y forma AUTOFAGOLISOSOMA Actúan enzimas acídicas e hidrolasas

16
Sistema Lisosomal Macroautofágico

17
Enfermedades asociadas a fallo en el sistema autofagolisosoma
Ataxia espinocerebelar Enfermedad de Parkinson familiar ( sinucleinopatías) Demencia frontotemporal
 Demencia frontotemporal.")
18
Apoptosis Es frecuente que se produzcan proteínas mal ensambladas o plegadas que activen el sistema de respuesta Soportan por corto tiempo proteínas defectuosas pero en condiciones crónicas se activa la APOPTOSIS

19
Apoptosis

20
Apoptosis La apoptosis esta afectada en las enfermedades neurodegenerativas Estas enfermedades son de inicio tardío Hace suponer que la incapacidad de eliminar moléculas anormales progresa con la edad Las proteínas acumuladas de forma intra o extraneuronal pueden dar apoptosis pero antes disfunción de la red neuronal

21
Enfermedades Neurodegenerativas
Inician el fenómeno fisiopatólogico muchos años antes del inicio de los síntomas El acúmulo de proteínas anormales no ocurre rápidamente, se acumulan antes de los síntomas Apoptosis esta afectada en la mayoría de enfermedades neurodegenerativas

22
CLASIFICACIÓN Combina descripción clínica, histológica y bioquímica
Clasificación general de las demencias degenerativas sinucleinopatias Enfermedad de Parkinson Atrofia multisistémica Demencia con cuerpos de Lewy Hallervorden-Spatz 2. Taupatías Degeneración corticobasal Parálisis supranuclear progresiva Demencia frontotemporal Enfermedad de gránulos argirófilos 3. Amiloidopatías Enfermedad de Alzheimer Angiopatía congofila 4. Poliglutaminopatías Enfermedad de Huntington Otras ataxias

23
Epidemiología Común en personas mayores
3 millones individuos con demencia en USA para el 2003 6-8% mayores de 65 30% mayores de 85 Vida media varía de acuerdo al diagnóstico una vez iniciado los síntomas.

24
Definición Llaves del diagnóstico:
Disfunción cognitiva, principalmente en memoria Interfiere significativamente en el trabajo o actividades sociales

25
Déficits Cognitivos En al menos dos o más de los siguientes:
En memoria reciente Lenguaje Función visuespacial Función ejecutiva Atención Orientación

26
Enfermedad de Alzheimer
La causa mas común de demencia en el adulto mayor 50-60% demostrado por autopsia La manifestación mas común: Olvido excesivo de las cosas

27
FISIOPATOLOGIA Cambios neuropatológicos:
Ovillos neurofibrilares Placas de amiloide ( placas seniles ) Neurotransmisores: Ach, NE, serotonina Genética: Polimorfismos Mutaciones esporádicas
 Neurotransmisores: Ach, NE, serotonina. Genética: Polimorfismos. Mutaciones esporádicas.")
28
Cambios Neuropatológicos
Áreas de Neocortex, hipocampo y corteza entorinal Depósitos extracelulares de B ‘amiloide en las placas seniles Formación intracelular de ovillos neurofibrilares: forma anormalmente fosforilada de proteína tau asociada a microtúbulos Pérdida de sinapsis neuronales y neuronas piramidales

29
Placas amiloides 1. Depósitos extracelulares de AA proteolíticos fragmentos derivados de una proteína transmembrana precursora de amiloide. 2.Células reactivas gliales ( fagocitos). 3.Componentes proteináceos: APO E y NAC ( componente no B amiloide), L sinucleinopatías y complemento.
. 3.Componentes proteináceos: APO E y NAC ( componente no B amiloide), L sinucleinopatías y complemento.")
30
Placas amiloides Depósitos existen predominantemente como AA de 42 péptidos Menos solubles Mayor agregación que 40 AA Vías de las secretasas: L secretasa: producto final ( P3) que no tiene placas amiloides B secretasa: producto final de 40 y 42 AA G secretasa : vía común La β-amiloide es un péptido de 36 a 43 aminoácidos que se sintetiza a partir de la Proteína precursora amiloide (APP). Aunque es generalmente conocida por su relación con la enfermedad de Alzheimer, no existe específicamente con ese fin. Se ha encontrado evidencia de que la β-amiloide tiene múltiples actividades no asociadas con la enfermedad.1
 que no tiene placas amiloides. B secretasa: producto final de 40 y 42 AA. G secretasa : vía común. La β-amiloide es un péptido de 36 a 43 aminoácidos que se sintetiza a partir de la Proteína precursora amiloide (APP). Aunque es generalmente conocida por su relación con la enfermedad de Alzheimer, no existe específicamente con ese fin. Se ha encontrado evidencia de que la β-amiloide tiene múltiples actividades no asociadas con la enfermedad.1.")
31
NO CORRELACIONAN CON GRADO DE DEMENCIA
Placas amiloides ¿PATOGNOMÓNICAS DE EA? NO Enfermedades por priones: Creutzfeld- Jakob Síndrome de Gerstmann-Sträussler-Sheinker Insomnio fatal familiar Demencia pugilística Cerebros de personas sin demencia clínica NO CORRELACIONAN CON GRADO DE DEMENCIA Los síntomas comienzan con una disartria que lentamente se desarrolla (dificultad para hablar) y ataxia cerebelosa (inestabilidad en la marcha). Luego viene la demencia progresiva que se hace más evidente. La pérdida de la memoria puede ser el primer síntoma que los familiares noten. Los hallazgos neuropatológicos incluyen la deposición generalizada de placas amiloides de proteína priónica anormalmente dobladas. enfermedad rara de carácter neurodegenerativo que afecta entre la tercera y la séptima década de vida. La incidencia exacta se desconoce pero se estima entre 1 y 10 por cada 100 millones. Casos entre parientes son asociados a consanguineidades. Parece que cambios genéticos son esenciales para la adquisición de la enfermedad 1986, AUTOSÓMICA dominante, anos, no pueden dormir, letargo, 8 m coma profundo, afectación de tálamo.
 y ataxia cerebelosa (inestabilidad en la marcha). Luego viene la demencia progresiva que se hace más evidente. La pérdida de la memoria puede ser el primer síntoma que los familiares noten. Los hallazgos neuropatológicos incluyen la deposición generalizada de placas amiloides de proteína priónica anormalmente dobladas. enfermedad rara de carácter neurodegenerativo que afecta entre la tercera y la séptima década de vida. La incidencia exacta se desconoce pero se estima entre 1 y 10 por cada 100 millones. Casos entre parientes son asociados a consanguineidades. Parece que cambios genéticos son esenciales para la adquisición de la enfermedad. 1986, AUTOSÓMICA dominante, anos, no pueden dormir, letargo, 8 m coma profundo, afectación de tálamo.")
32
Ovillos neurofibrilares
1.Agregados intracelulares relativamente insolubles 2.Filamentos de una proteína (tau) hiperfosforilada anormalmente asociada a microtúbulos 3.Proteína tau: ensamblaje y estabilización de microtúbulos 4.Acumulación desestabilización y disrupción del citoesqueleto del sistema transportador intracelular
 hiperfosforilada anormalmente asociada a microtúbulos. 3.Proteína tau: ensamblaje y estabilización de microtúbulos. 4.Acumulación desestabilización y disrupción del citoesqueleto del sistema transportador intracelular.")
33
Ovillos neurofibrilares
¿PATOGNOMÓNICOS DE EA? NO 1. Enfermedades neurodegenerativas: Demencia frontotemporal Degeneración córticobasal Parálisis supranuclear progresiva 2. Demencia pugilística 3.Cerebros de personas sin demencia clínica CORRELACIONA CON EL GRADO DE DEMENCIA

34
Relación ON - PS Evento inicial
Alteraciones en la proteína precursora de amiloide Acumulación intraneural de B amiloide Disrupción intracelular de vías de transporte Pérdida de proteína precursora de amiloide soluble presináptica ( B amiloide 42 ) Aumenta fosforilación de Tau, disrupción de la integridad del sistema microtubular
 Aumenta fosforilación de Tau, disrupción de la integridad del sistema microtubular.")
35
Relación ON-PS Acumulación extracelular de remanentes de neuronas degeneradas – B amiloide Precipitan eventos patológicos secundarios Activación de microglia – Respuesta inflamatoria LESION CEREBRAL

36
Genética 1. Mutaciones: 2. Polimorfismos genéticos: Causa menos común
1o mutaciones en el gen de la proteína precursora de amiloide---cromosoma 21 60 mutaciones en el gen de presenilina 1 (PS1)---- cromosoma 14 2 mutaciones gen presenilina 2 ( PS2)---cromosoma 1 Presenilina: proteína transmembrana importante en el clivaje de PPA por la B- secretasa y G- secretasa 2. Polimorfismos genéticos: Alelo E4 de ApoE
---- cromosoma mutaciones gen presenilina 2 ( PS2)---cromosoma 1. Presenilina: proteína transmembrana importante en el clivaje de PPA por la B- secretasa y G- secretasa. 2. Polimorfismos genéticos: Alelo E4 de ApoE.")
37
NEUROTRANSMISORES SISTEMA COLINERGICO
1914, Henry Dale CAT NT modulador más importante del cerebro ACE –BuCE ( amígdala e hipocampo ) Investigaciones BQ en Receptores : Muscarínicos y nicotínicos 1974, David Drachman utilizó escopolamina reproduciendo en sujetos sanos déficit de memoria 95% disminución actividad colinérgica: Areas 20,21,22,28 Bradman Colina – acetil CoA
 Investigaciones BQ en Receptores : Muscarínicos y nicotínicos. 1974, David Drachman utilizó escopolamina reproduciendo en sujetos sanos déficit de memoria. 95% disminución actividad colinérgica: Areas 20,21,22,28 Bradman. Colina – acetil CoA.")
38
SISTEMA COLINERGICO Sistema cerebro basal anterior
Encargado de mantener operativa la corteza cerebral y papel decisivo en procesos de memoria y atención selectiva. Bucle córtico estriatal controla percepción,aprendizaje, conocimiento, afectividad, juicio y sueño REM 1. Núcleos prosencéfalo basal: Ach 2. Núcleos del rafé: serotonina 3. Locus coeruleus: NA-A 4. Sustancia nigra: DA

39
SISTEMA COLINERGICO 3 sistemas:
1. Inervación HIPOCAMPO, amígdala, córtex frontal, parietal, temporal y occipital. Núcleo de MEYNERT: CAT y ACE 2.Tronco cerebral, parte rostral: sistema pedúnculo-pontino-tálamo-córtico-retículo- nigral 3.Estriado: ganglios de base – corteza frontal

40
Sistema serotoninérgico
1-2 % se produce en el cerebro ( regiones del Rafé del tronco cerebral, protuberancia) por hidroxilación del triptófano y posterior descarboxilación Resto se produce en plaquetas, mastocitos y células cromafines NT inhibitorio Disminución actividad colinérgica Alteración de receptores postsinápticos de 5-HT secundario a denervación colinérgica
 por hidroxilación del triptófano y posterior descarboxilación. Resto se produce en plaquetas, mastocitos y células cromafines. NT inhibitorio. Disminución actividad colinérgica. Alteración de receptores postsinápticos de 5-HT secundario a denervación colinérgica.")
41
Sistema noradrenérgico
Cerebro, células cromafines, ganglios y nervios simpáticos Locus coeruleus y tegmento lateral Inhibidor Aumento de placas seniles, disminución de la actividad de la DBH 40% disminución actividad DBH Alteración tardía

42
Sistema dopaminérgico
Actividad motora, emoción y motivación Cambios no son constantes Síntomas extrapiramidales asociados y no tanto con la cognición

43
Enfermedad de Alzheimer
La observación inicial es que el paciente repite preguntas Olvida pagar recibos, tomarse tratamientos, y tiene problemas de orientación Apatía y desinterés por actividades que realizaba previamente Algunos cursan con depresión severa

44
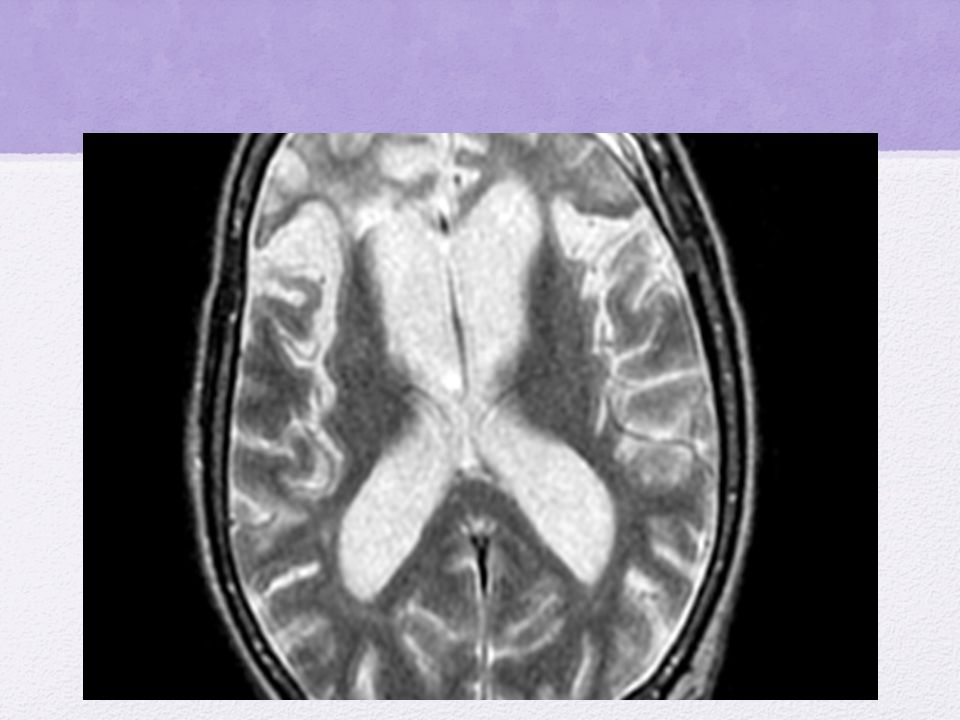
Enfermedad de Alzheimer
Estudios Diagnostico: RMN muestra atrofia del hipocampo Determinación de Beta amiloide y proteína tau en LCR otros… Pronóstico malo Sobrevida en relación a estadio de dx Tratamiento: inhibidores de acetilcolinesterasa

45
Demencia Asociada a Parkinsonismo Demencia por cuerpos de Lewy
Cuerpos de Lewy presentes en autopsia en 10-30% Parkinsonismo espontáneo precede a la demencia por varios años Pacientes desarrollan manifestaciones motoras y trastornos del sueño Trastorno cognitivo asociado a amnesia anterógrada Mutación L – sinucleína y cromosoma 4 ( forma familiar) Disminución actividad Ach y DA
 Disminución actividad Ach y DA.")
46
Demencia por cuerpos de Lewy
1912 Clásicamente asociadas a Enfermedad de Parkinson primaria Inclusiones citoplasmáticas en neuronas colinérgicas y monoaminas Sustancia nigra, locus ceruleus y núcleo basal de Meynert Centro denso eosinofílico rodeado de halo claro

48
Demencia Asociada a Parkinsonismo Demencia por cuerpos de Lewy
Trastornos motores: Trastorno de la marcha Balance Rigidez Bradiquinesia Riesgo de caídas

49
Demencia Asociada a Parkinsonismo Demencia por cuerpos de Lewy
Trastorno cognitivo: Predominantemente amnesia anterógrada Con mayor deterioro en función ejecutiva y visuespacial

50
Demencia Asociada a Parkinsonismo Demencia por cuerpos de Lewy
Trastorno del sueño: REM Sueño excesivo durante el día Alucinaciones visuales muy bien estructuradas Sueños vívidos

51
Demencia Asociada a Parkinsonismo Demencia por cuerpos de Lewy
Tienen progresión mas acelerada que la EA y sobrevida más corta Con mejoría ante tratamiento para disfunción motora, trastorno del sueño, y deterioro cognitivo usando inhibidores de acetilcolinesterasa

52
Demencia Frontotemporal
Maurice Ravel

53
Demencia frontotemporal
Presentación dramática semejante a trastorno psiquiátrico Principales manifestaciones son cambios en la personalidad, comportamiento y juicio Preservación relativa de la memoria El TAC es útil en el diagnóstico: Atrofia focal prefrontal o atrofia temporal anterior

55
Demencia Frontotemporal
1892 Arnold Pick : atrofia lobar con demencia afásica 1911 Louis Alzheimer describe los cuerpos de Pick Inclusiones citoplasmáticas de proteína tau Variante de FTD con cuerpos de Pick es rara

56
Demencia Frontotemporal
Representa 50% de las demencias en menores de 60 años 5% de todas las demencias neurodegenerativas Esporádica la mayoría Mutación genética: Proteína tau cromosoma 18 Tres variantes: Demencia frontotemporal: lóbulo frontal Afasia progresiva no fluente: lóbulo frontal izq Demencia semántica: lóbulo temporal izq

57
Demencias Rápidamente Progresivas Enfermedad de Creutzfeldt-Jacob
Demencia de inicio subagudo (semanas o meses) Acompañado de manifestaciones motoras como extrapiramidalismo, síndromes extraoculares o cerebelares. Inicia con trastornos de conducta y personalidad (depresión y agitación) Deterioro cognitivo Convulsiones frecuentes en el progreso de la enfermedad Forma más frecuente es la esporádica 15% familiares: autosómica dominante, gen PRND cromosoma 20
 Acompañado de manifestaciones motoras como extrapiramidalismo, síndromes extraoculares o cerebelares. Inicia con trastornos de conducta y personalidad (depresión y agitación) Deterioro cognitivo. Convulsiones frecuentes en el progreso de la enfermedad. Forma más frecuente es la esporádica. 15% familiares: autosómica dominante, gen PRND cromosoma 20.")
58
Enfermedad de Creutzfeldt-Jacob
Encefalopatía espongiforme más común del ser humano 1980 ¨enfermedad de las vacas locas¨ Scrapie ( ovejas y cabras ) Prusiner: moléculas compuestas únicamente por material proteico Priones capaces de propagarse en un mismo huésped y trasmitirse a otros en períodos de incubación prolongados Proteína priónica es solamente un receptor para un virus No se conoce mecanismo exacto de propagación
 Prusiner: moléculas compuestas únicamente por material proteico. Priones capaces de propagarse en un mismo huésped y trasmitirse a otros en períodos de incubación prolongados. Proteína priónica es solamente un receptor para un virus. No se conoce mecanismo exacto de propagación.")
60
Enfermedad de Huntington
Autosómica dominante Etapa media de la vida adulta Espectativa de vida años Inicialmente coreoatetosis y luego demencia Perturbaciones emocionales, trastornos de la conducta y la personalidad

61
Enfermedad de Huntington
Pérdida de neuronas en los núcleos caudados y gliosis Repetición del trinucleótido CAG ( excesivamente largo), brazo corto del cromosoma 4 Acumulación intraneuronal de huntingtina (producto anormal de la secuencia CAG )
, brazo corto del cromosoma 4. Acumulación intraneuronal de huntingtina (producto anormal de la secuencia CAG )")
62
Enfermedad de Huntington

63
Conclusiones Las enfermedades neurodegenerativas son llamadas proteinopatias porque su origen ocurre en la acumulación de alguna proteína y su eliminación depende de varios sistemas de desintegración proteica

64
Conclusiones El fallo en los mecanismos de eliminación de proteínas anormales es el factor inicial o acelerante de las enfermedades neurodegenerativas

65
Conclusiones Las enfermedades neurodegenerativas son de inicio tardío lo que hace suponer que la incapacidad de eliminar moléculas anormales progresa con la edad

Presentaciones similares






>")














