Descargar la presentación
La descarga está en progreso. Por favor, espere

1
Pilar de león Janeth Montenegro Danysabel pitti
Misión n°1 y lisosomas Pilar de león Janeth Montenegro Danysabel pitti

2
Misión n° 1 La misión numero uno se traba de controlar y dirigir el nanobot recolector. Con el teníamos que rescatar los nanobort averiados en varios lugares dé la célula y había que tener cuidado con los lisosomas porqué ellos digieren cualquiera cosa que o debe estar en la célula y al final se recogían cuatro y se llevaban al centro de comando.

4
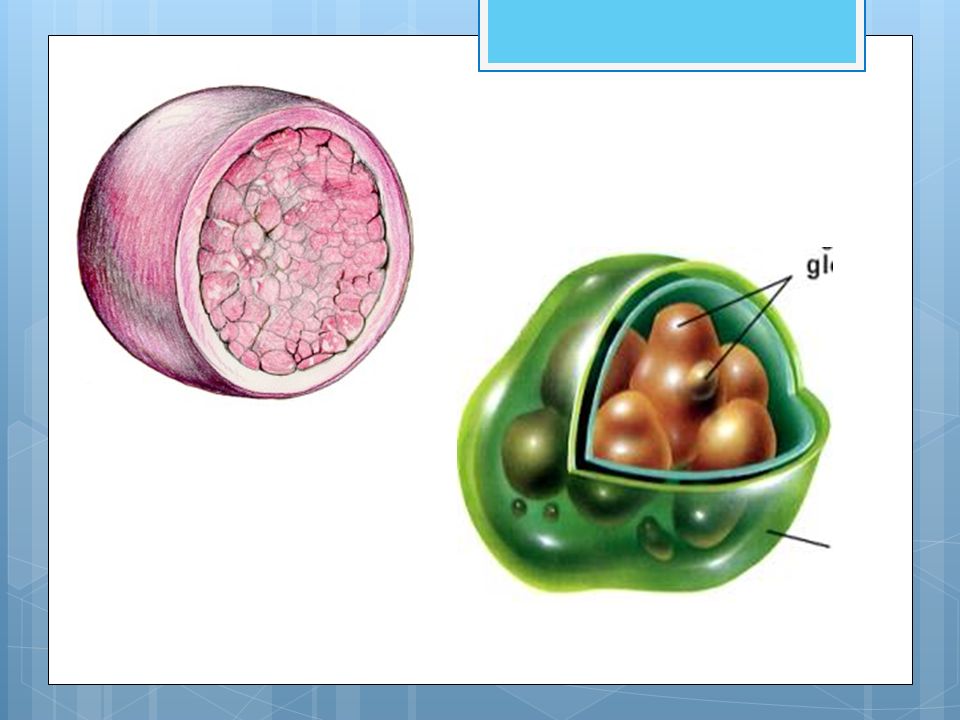
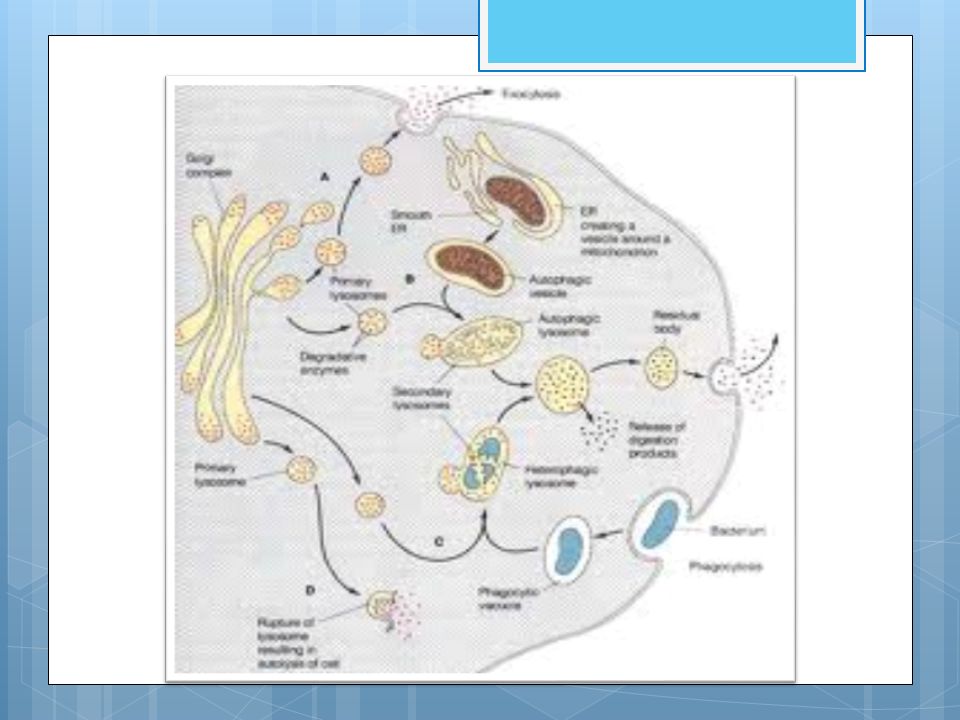
Los lisomas Los lisosomas son orgánulos relativamente grandes, formados por el retículo endoplasmático rugoso y luego empaquetadas por el complejo de Golgi, que contienen enzimas hidrolíticas y proteolíticas que sirven para digerir los materiales de origen externo (heterofagia) o interno (autofagia) que llegan a ellos. Es decir, se encargan de la digestión celular.
 o interno (autofagia) que llegan a ellos. Es decir, se encargan de la digestión celular.")
6
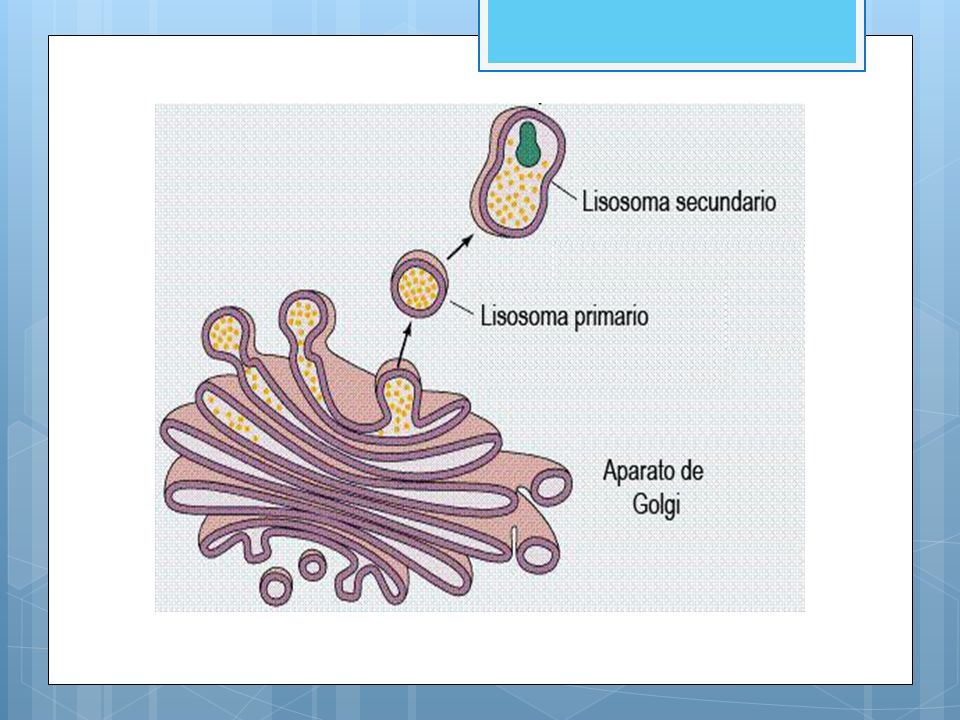
Se diferencian dos tipos de lisosomas:
Lisosomas primarios: son los que salen de las cisternas del aparato de Golgi, contienen enzimas digestivas y todavía no han participado en la digestión. Lisosomas secundarios: se forman al unirse con otras vesículas, por lo que han realizado la digestión.

8
Funciones lisosomales:
Los lisosomas participan en la muerte celular. No participan en el desarrollo embrionario, pero si intervienen en el proceso de diferenciación de órganos durante la ontogenia. Intervienen en la digestión de las sustancias ingeridas por endocitosis. Éstas vacían su contenido en endosomas, y la fusión de un endosoma con un lisosoma primario forma un lisosoma secundario.

9
Las enzimas lisosómicas y atienen acceso a un sustrato
Las enzimas lisosómicas y atienen acceso a un sustrato. En el caso de la fagocitosis, los fagosomas también se unen con lisosomas primarios para dar secundarios Los productos no degradados quedan en un cuerpo rodeado de membrana que pueden ser defecados por unión de la membrana de la vacuola.

11
¿Que pasaría si los lisosomas fallan en el reconocimiento de que digerir y que no?
Si fallan los Lisosomas la célula no puede realizar las funciones de Digestión Intracelular o Endocitosis, en este caso la masa alimenticia incorporada por la membrana plasmática en forma de una Vacuola alimenticia no podría ser reducida en metabolitos intermedios por falla de estos organelos, lo mismo que la eliminación de una Noxa biológica(Virus, Bacterias) que pudiesen entrar en el interior de la célula, por otro lado no pdría realizar la Exocitosis, en este caso los Lisosomas fusionan su membrana biológica con la membrana palsmática para realizar la Digestión extracelular protegiendo a la célula de la invasión de noxas biológicas.
 que pudiesen entrar en el interior de la célula, por otro lado no pdría realizar la Exocitosis, en este caso los Lisosomas fusionan su membrana biológica con la membrana palsmática para realizar la Digestión extracelular protegiendo a la célula de la invasión de noxas biológicas.")
12
Lisomas cuando fallan en el reconocimiento

13
Enfermedades lisosómicas
Son enfermedades causadas por la disfunción de algún enzima o por la liberación incontrolada de dichas enzimas en el citosol, lo que produce la lisis celular. Esfingolipidosis. Son enfermedades causadas por la disfunción de alguna de las enzimas de la ruta de degradación de los esfingolípidos. La Enfermedad de Gaucher es una enfermedad metabólica rara, que se debe a la deficiencia de la enzima betaglucosidasa ácida, que origina un depósito de un glucocerebrósido, la glucosilceramida, en el sistema retículoendotelial.

14
Gaucher Tipo I, del adulto o no neuroléptica, Tipo II, aguda neuroléptica, o cerebral infantilTipo III, neuroléptica subaguda,

15
Enfermedad de Niemann-Pick
Enfermedad de Niemann-Pick. Esta enfermedad genética se presenta bajo cuatro formas conocidas como tipos A, B, C y D. Cada una involucra diferentes órganos y puede inutilizar áreas tan básicas del organismo como el sistema nervioso central o el aparato respiratorio

16
La enfermedad de Wolman es una lipidosis congénita, que se transmite de una forma autosómica recesiva y se debe al déficit de una enzima lisosómica que es codificada en el brazo largo del cromosoma 10: la lipasa ácida. enfermedad de Pompe. Es un defecto de la α(1-4) glucosidasa ácida lisosómica, también denominada maltasa ácida. El glucógeno aparece almacenado en lisosomas.
 glucosidasa ácida lisosómica, también denominada maltasa ácida. El glucógeno aparece almacenado en lisosomas.")
17
enfermedad de Hurler: es una enfermedad metabólica hereditaria muy rara, alcanza una incidencia de 1 en 100,000 a nivel mundial; si se considera esta cifra como correcta, hasta el momento estos casos pasaban desapercibidos, o los pacientes fallecían sin llegar a establecerse el diagnóstico definitivo de la entidad.

Presentaciones similares