Descargar la presentación
La descarga está en progreso. Por favor, espere

1
Fisiopatología del Sistema Endocrino
UNIBE Dr. José R. Fuchs C.

2
Alteraciones del Sistema Endocrino
DEFICIENCIA HORMONAL EXCESO DE HORMONAS RESISTENCIA DE ACCION A LAS HORMONAS ESTADOS POR DEFICIENCIA HORMONAL: Procesos destructivos Infecciones ( Tuberculosis adrenal) Infartos (Sheehan) Inflamación ( DM secundaria a pancreatitis) Tumores (células nulas de la pituitaria) Autoinmune (tiroiditis de Hashimoto) Hereditarias (enanismo pituitario) EXCESO DE HORMONAS: Sobreproducción de la glándula Sobrestimulación de la glándula Producida por tejido extraglandular Iatrogénica Destrucción de la glándula blanco
 Infartos (Sheehan) Inflamación ( DM secundaria a pancreatitis) Tumores (células nulas de la pituitaria) Autoinmune (tiroiditis de Hashimoto) Hereditarias (enanismo pituitario) EXCESO DE HORMONAS: Sobreproducción de la glándula. Sobrestimulación de la glándula. Producida por tejido extraglandular. Iatrogénica. Destrucción de la glándula blanco.")
3
Alteraciones del Sistema Endocrino
RESISTENCIA HORMONAL Generalmente de carácter hereditario Deficiencia en receptores o post-recepción Desarrollo de anticuerpos a las hormonas o sus receptores Ausencia de células blanco PRODUCCION DE HORMONAS ANORMALES Algunos tipos de DM Inmunoglobulinas que se fijan al receptor específico y lo estimulan Anticuerpos anti-receptores de insulina. DEFECTOS QUE AFECTAN SISTEMAS ENDOCRINOS MULTIPLES Panhipopituitarismo Sindromes poliglandulares Sindromes de neoplasias endocrinas múltiples

4
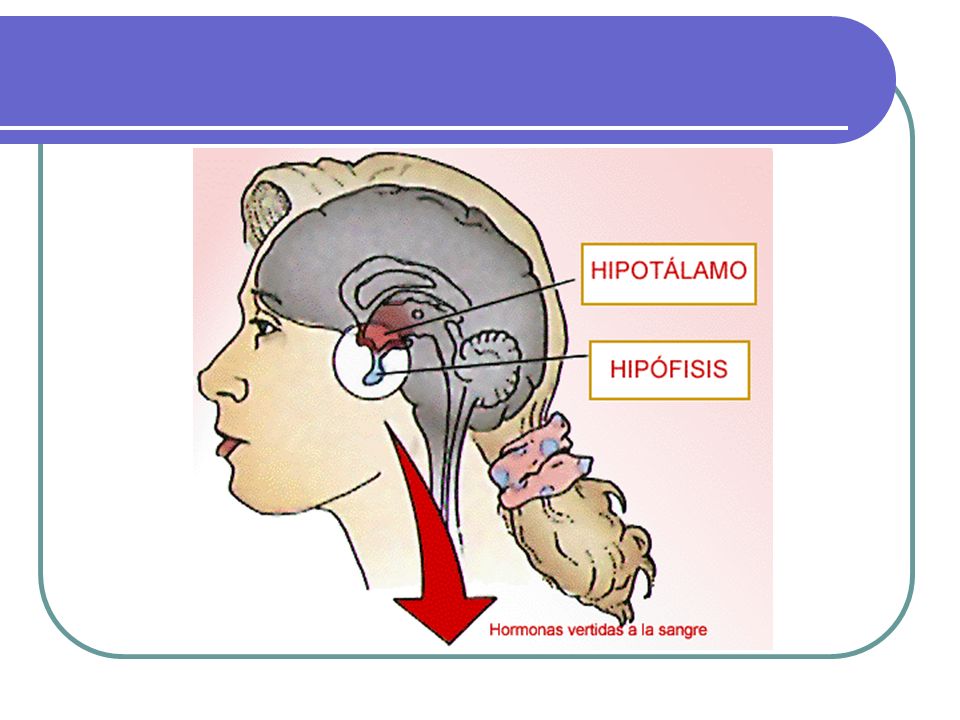
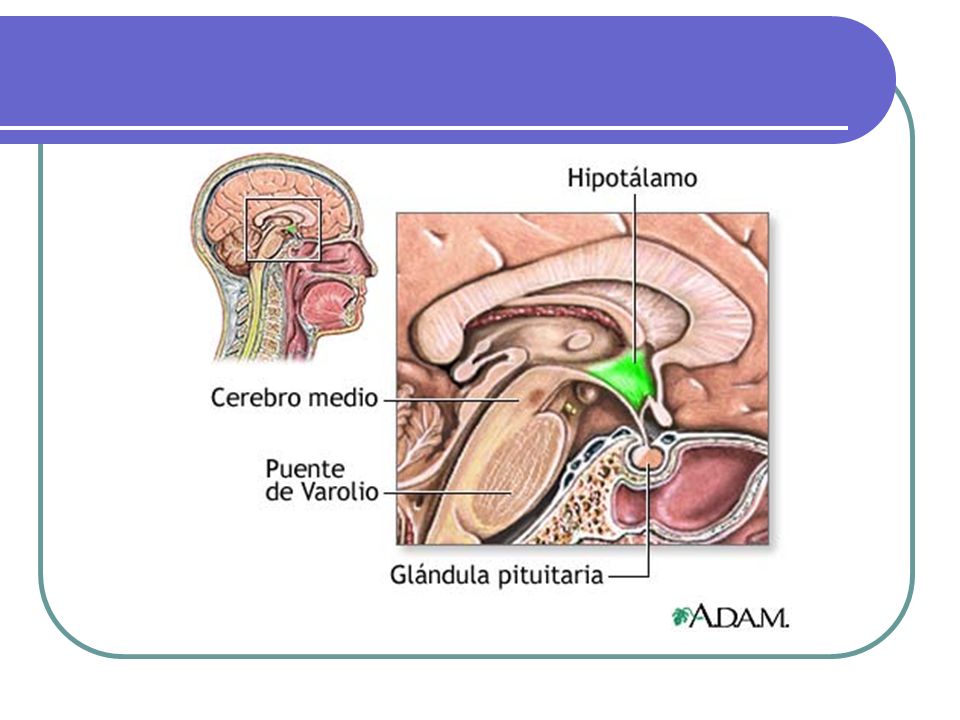
HIPOTÁLAMO

7
HIPOTÁLAMO HIPOTALAMO Funciones no endocrinas: Funciones endocrinas:
Peso: 4 g. Enfermedad ocurre sólo cuando el compromiso es bilateral. . Funciones no endocrinas: Apetito y patrones alimentarios Regulación de la temperatura Ciclo sueño-vigilia Memoria y comportamiento Sed Función del sistema autonómico. Funciones endocrinas: Factores liberadores de hormonas estimulantes hipofisiarias: CRH; TRH; GHRH; GnRH. Dos sustancias inhibidoras: Somatostatina a la GH y la dopamina a la prolactina. Además de ocitocina y vasopresina u hormona antidiurética.

9
HIPOTÁLAMO TUMORES. MANIFESTACIONES CLÍNICAS:
Tumores son de crecimiento lento y usualmente tienen que alcanzar gran tamaño para producir sintomatología: Hidrocefalia por compromiso del tercer ventrículo Disfunción hipotalámica endocrina y no endocrina Hipopituitarismo parcial o total Demencia Trastornos en la ingesta de alimentos (obesidad, emación) Disfunción endocrina Los procesos agudos o de crecimiento rápido: Coma Alteraciones en la función autonómica
 Disfunción endocrina. Los procesos agudos o de crecimiento rápido: Coma. Alteraciones en la función autonómica.")
10
HIPOTÁLAMO Lesiones en el hipotálamo anterior:
Craneofaringioma Gliomas del nervio óptico Meningiomas Enf granulomatosas Germinomas Aneurismas de la carótida interna Lesiones del hipotálamo posterior: Gliomas Hamartomas Ependimomas Teratomas

11
Hipotálamo CRANEOFARINGIOMAS
3-5% de todas las neoplasias intracraneanas. Mayoría son supracelares, 15% intracelares. Frecuentemente quísticas y calcificadas. La mayoría se manifiestan en la infancia. 45% en > de 20 años y 20% en > de 40 años. CLINICA: Hipertensión endocraneana: cefalea, vómitos y papiledema. Trastornos visuales, hemianopsias. Baja estatura. En adultos el 80% tendrán trastornos visuales, cefalea, deterioro mental y cambios de conducta, hipogonadismo e hiperprolactinemia (50%). Menos frecuente: Diabetes insípida, obesidad, panhipopituitarismo.
. Menos frecuente: Diabetes insípida, obesidad, panhipopituitarismo.")
12
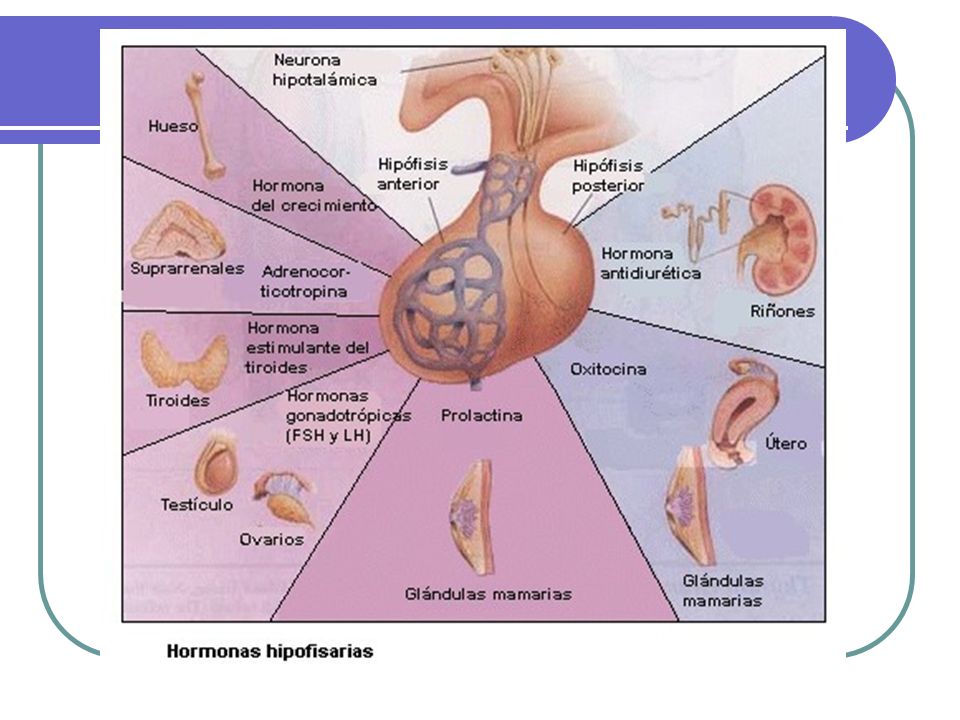
HIPÓFISIS

15
Hipófisis TUMORES DE LA HIPOFISIS
10 al 15% de todas los tumores intracraneanos Microadenomas < 1cm diámetro. Macroadenomas > 1cm. Usualmente se diagnostican por: Rx u otros estudios de imágenes de cráneo. Defectos visuales, cefalea u otros defectos neurológicos, especialmente con macroadenomas Datos de producción hormonal en exceso: hiperprolactinemia, cushing, acromegalia Hipopituitarismo debido a destrucción de la región hipotálamo hipofisiaria. Apoplejía pituitaria.

16
HIPÓFISIS TIPOS DE TUMORES Porlactinoma: micro o macroadenoma.
Secretores de HC: macroadenomas generalmente Enfermedad de Cushing: microadenomas productores de ACTH muy pequeños que a veces no se detectan por TAC O RM. Adenomas cromófobos, incidentalomas o de células nulas; no producen hormonas. (30-40%) (los más frecuentes) MUY PERO MUY RAROS Secretores de (TSH). (EXCEPCIONALES) Secretores de (FSH y LH): hipogonadismo, problemas visuales y cefaleas. (MUY RAROS)
 (los más frecuentes) MUY PERO MUY RAROS. Secretores de (TSH). (EXCEPCIONALES) Secretores de (FSH y LH): hipogonadismo, problemas visuales y cefaleas. (MUY RAROS)")
17
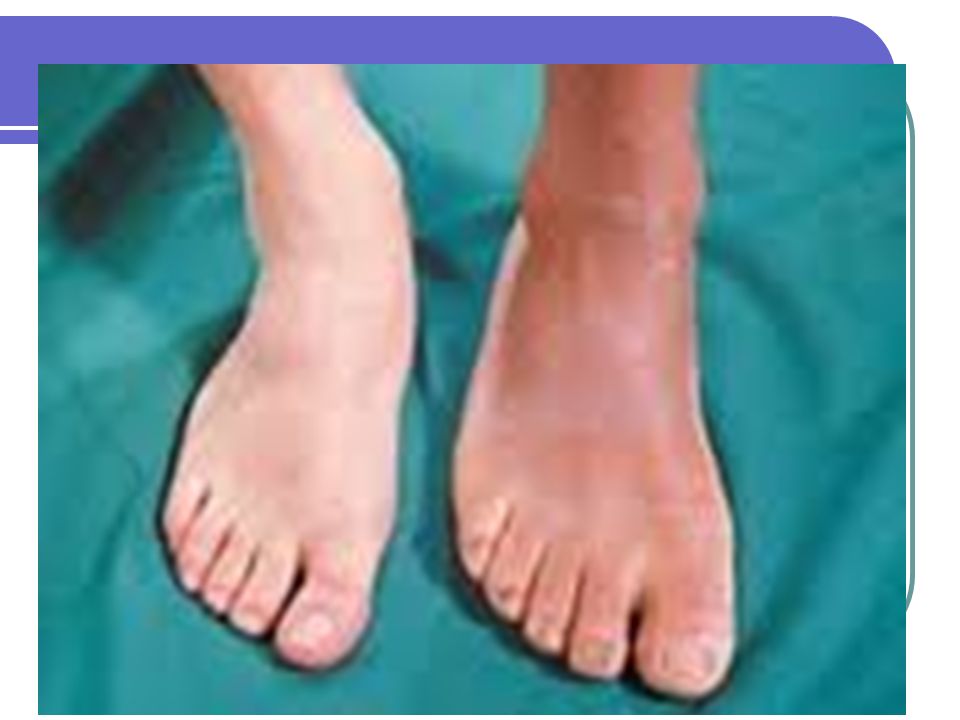
Adenomas hipofisiarios
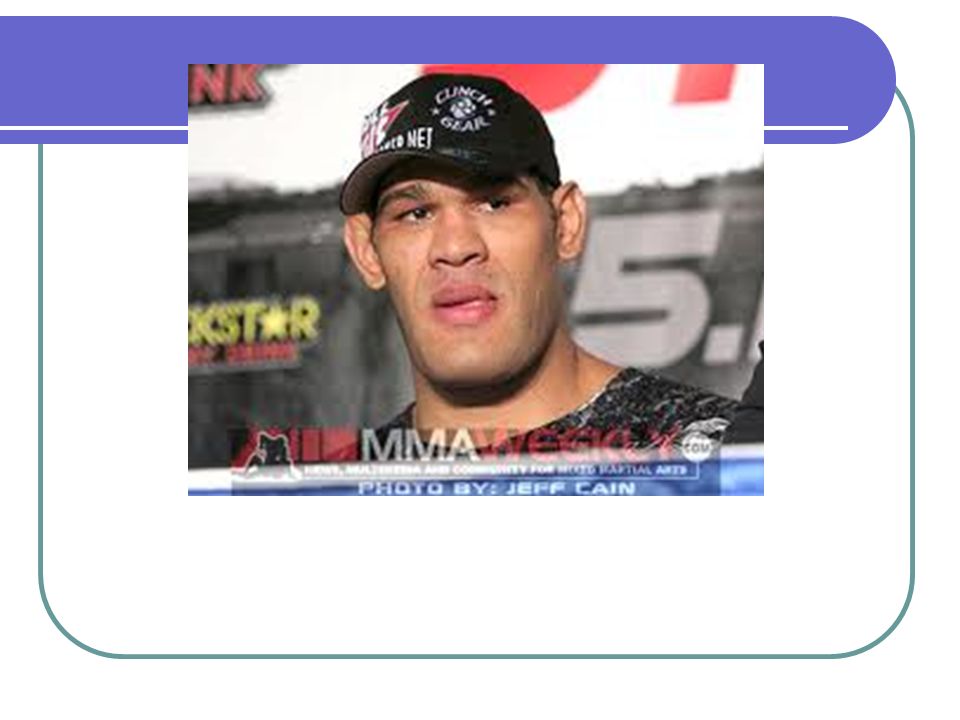
Acromegalia: Exceso de hormona crecimiento Hipotálamo---GHRF est (+). Somatostatina inh (-) la producción de GH por la hipófisis : GH estimula la producción de somatomedinas (c) (ILGF) factor de crecimiento similar a la insulina, en el hígado Mayoría por macroadenomas hipofisiarios. 40 casos por millón. Raro por aumento de GHRF <1%. Producción ectópica de GH es rara: Carcinoma bronquial de células pequeñas Carcinoma medular de tiroides Carcinoides del intestino delgado
. Somatostatina inh (-) la producción de GH por la hipófisis : GH estimula la producción de somatomedinas (c) (ILGF) factor de crecimiento similar a la insulina, en el hígado. Mayoría por macroadenomas hipofisiarios. 40 casos por millón. Raro por aumento de GHRF <1%. Producción ectópica de GH es rara: Carcinoma bronquial de células pequeñas. Carcinoma medular de tiroides. Carcinoides del intestino delgado.")
18
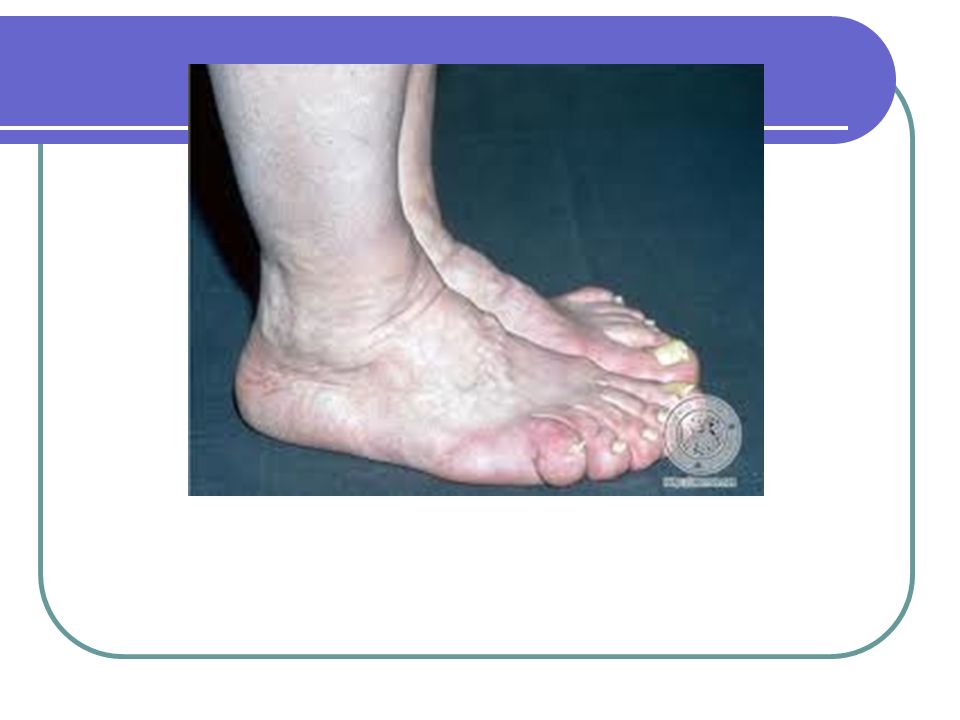
Crecimiento de las manos

24
CAUSAS DE HIPERPROLACTINEMIA
ESTADOS FISIOLOGICOS: A Embarazo B Lactancia C Estrés E Estimulación del pezón F Ingesta de alimentos MEDICAMENTOS: A. Antagonistas de receptores dopaminérgicos Fenotiazinas Butirofenonas Tioxaminas Metoclopramida Sulpiride B Agentes que depletan dopamina Metildopa Reserpina C Estrógenos D Opiáceos

25
Causas de hiperprolactinemia
ENFERMEDADES ASOCIADAS: A Tumores pituirtarios: Prolactinomas Adenomas productores de GH y prolactina Adenomas secretores de ACTH y prolactina Adenomas cromófobos no funcionantes B Enfermedad hipotalámica y del pedículo hipofisiario Enfermedad granulomatosa (sarcoidosis) Craniofaringiomas y otros tumores Irradiación craneana Sección del pedículo Silla vacía Anormalidades vasculares (aneurismas) Hipofisitis linfocítica Carcinoma metastásico C Hipotiroidismo primario D Insuficiencia renal crónica E Cirrosis F Trauma Tórax (cirugía, Herpes zoster) G Convulsiones
 Craniofaringiomas y otros tumores. Irradiación craneana. Sección del pedículo. Silla vacía. Anormalidades vasculares (aneurismas) Hipofisitis linfocítica. Carcinoma metastásico. C Hipotiroidismo primario. D Insuficiencia renal crónica. E Cirrosis. F Trauma Tórax (cirugía, Herpes zoster) G Convulsiones.")
26
Hipopituitarismo CLASIFICACION DE HIPOPITUITARISMO: AISLADO:
Deficiencia congénita de GNRH (hormona liberadora de gonadotrofinas). Deficiencia gonadal, ausencia de pubertad, bajos niveles plasmáticos de LH y FSH; se asocia a anosmia. Deficiencia aislada y adquirida de TSH: hipotiroidismo, pobre respuesta de TSH a TRH, se puede ver en la apoplejía pituitaria. Deficiencia aislada de ACTH, causando deficiencia selectiva de glucocorticoides, ausencia de hiperpigmentación, o contracción de volumen. El cortisol plasmático está bajo y no se eleva después de hipoglicemia inducida por insulina. Deficiencia aislada de hormona del crecimiento (GH). Estatura baja Todas estas deficiencias aisladas de las hormonas hipofisiarias son sumamente raras.
. Deficiencia gonadal, ausencia de pubertad, bajos niveles plasmáticos de LH y FSH; se asocia a anosmia. Deficiencia aislada y adquirida de TSH: hipotiroidismo, pobre respuesta de TSH a TRH, se puede ver en la apoplejía pituitaria. Deficiencia aislada de ACTH, causando deficiencia selectiva de glucocorticoides, ausencia de hiperpigmentación, o contracción de volumen. El cortisol plasmático está bajo y no se eleva después de hipoglicemia inducida por insulina. Deficiencia aislada de hormona del crecimiento (GH). Estatura baja. Todas estas deficiencias aisladas de las hormonas hipofisiarias son sumamente raras.")
27
Hipopituitarismo PANHIPOPITUITARISMO:
Más común que las deficiencias aisladas. Destrucción de la pituitaria por un tumor, craniofaringioma o metastásico, tumores de la región hipotálamo-hipofisiaria. Síndrome de Sheehan o infarto pituitario post-parto: ausencia de lactación, bajos niveles de prolactina, ausencia de respuesta a TRH, insuficiencia gonadal, hipotiroidismo, insuficiencia suprarrenal y otras. Otras causas: enfermedades granulomatosas (ej. sacoidosis, tuberculosis, granulomatosis de Wegener), infecciones, radioterapia.
, infecciones, radioterapia.")
28
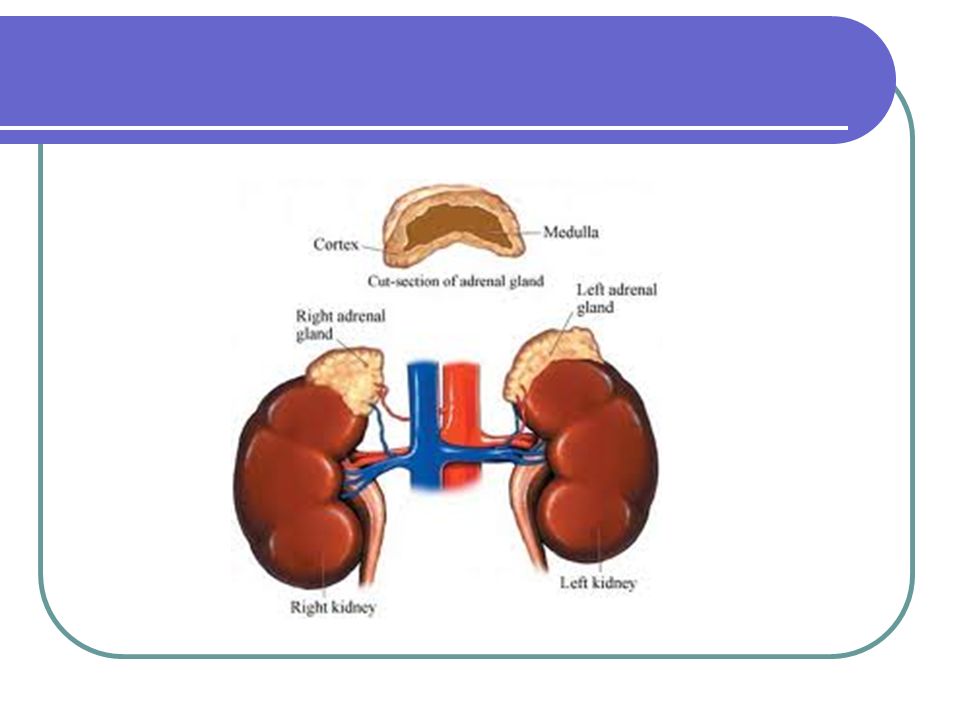
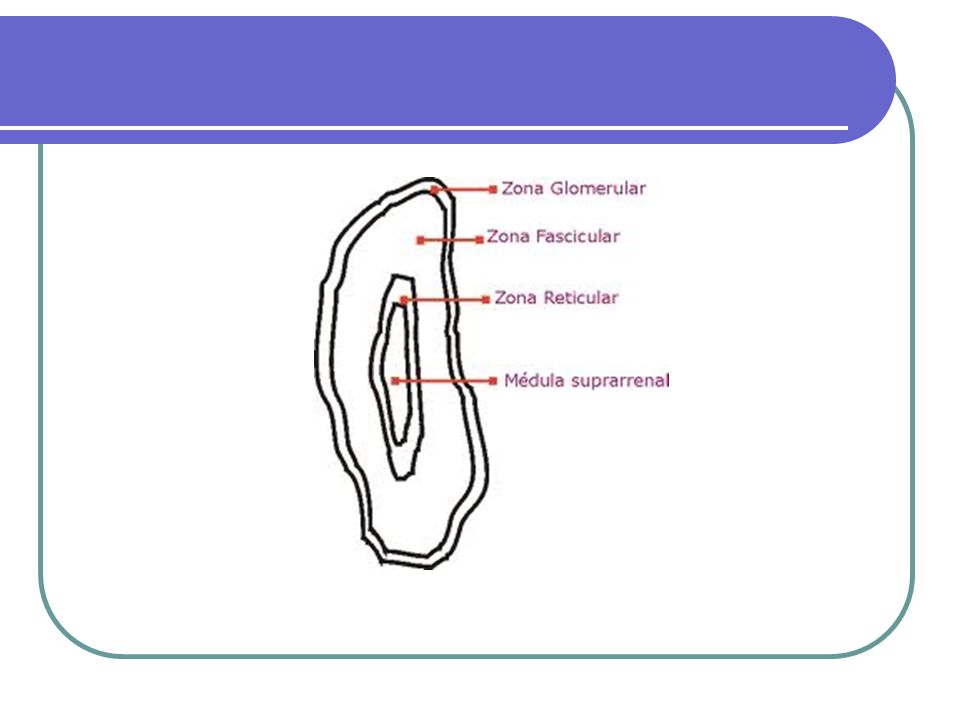
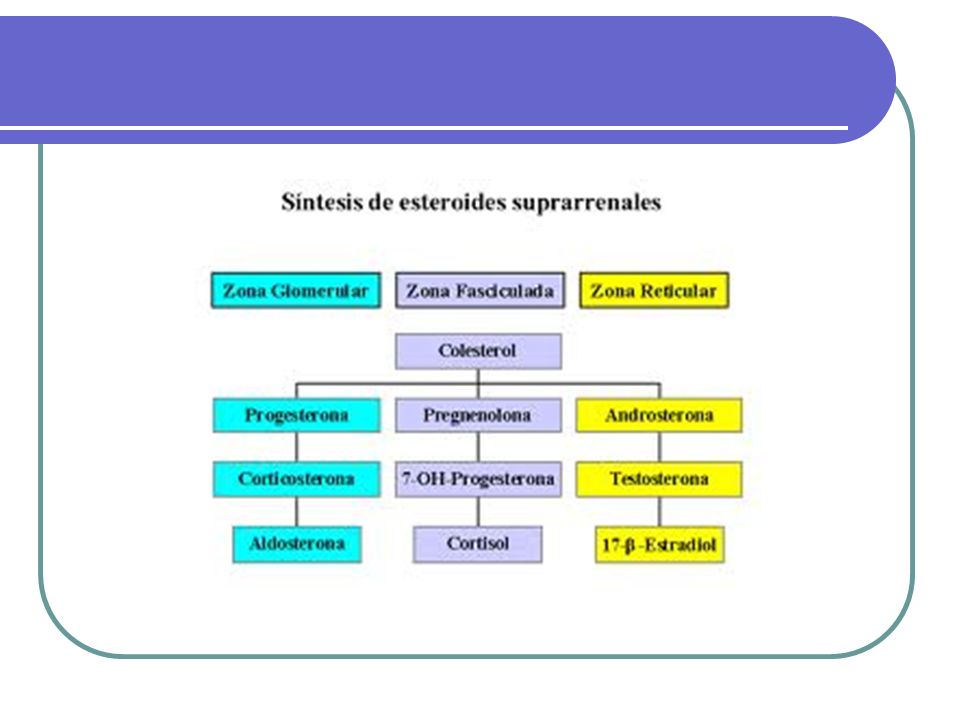
Fisiopatología de la glándula adrenal
UNIBE Dr. José R. Fuchs

33
Síndrome de Cushing La causa más común de Síndrome de Cushing es la iatrogénica debido a la administración de dosis terapéuticas de glucocorticoides. CAUSAS DE SINDROME DE CUSHING CAUSAS INCIDENCIA RELATIVA Dependientes de ACTH 75% Enfermedad de Cushing 60% Secreción ectópica de ACTH 15% Secreción ectópica de CRH Rara Independientes de ACTH Cáncer adrenal 25% Adenoma adrenal 10% Enfermedad adrenal micronodular Rara Ficticia o iatrogénica Muy común

34
Síndrome de Cushing TUMORES ASOCIADOS CON EL SINDROME DE ACTH ECTOPICO
Carcinoma de células de avena 50% Tumores embrionarios 35% Carcinoma del timo Carcinoma islotes Carcinoma medular de la tiroides Carcinoide bronquial Feocromocitoma 5% Otros % Gonadas Carcinoma de la próstata y del cervix Tumores de origen desconocido

35
Enfermedad de Cushing La enfermedad de Cushing (producción de ACTH por un microadenoma pituitario) es la causa más frecuente del Síndrome de Cushing. Es 4 veces más frecuente en mujeres que en hombres y a menudo ocurre entre las edades de 20 a 40 años. Usualmente es muy pequeño, sin embargo una TAC o una RM son capaces de detectarlos en un 50% de los casos.

36
Síndrome de Cushing Los tumores adrenales, causando un Síndrome de Cushing, se pueden presentar a cualquier edad, pero predominan en niños (particularmente carcinoma). Los carcinomas son usualmente muy agresivos y grandes y pueden palparse en la mayoría de los casos; en contraste los adenomas son de crecimiento lento y de tamaño moderado (1,5 a 6 cm de diámetro al diagnóstico), rara vez se asocian al MEN I. La hiperplasia adrenal micronodular ocurre en niños y adultos jóvenes, la patogénesis bioquímica no se conoce con certeza, puede ocurrir esporádicamente como enfermedad familiar.
. Los carcinomas son usualmente muy agresivos y grandes y pueden palparse en la mayoría de los casos; en contraste los adenomas son de crecimiento lento y de tamaño moderado (1,5 a 6 cm de diámetro al diagnóstico), rara vez se asocian al MEN I. La hiperplasia adrenal micronodular ocurre en niños y adultos jóvenes, la patogénesis bioquímica no se conoce con certeza, puede ocurrir esporádicamente como enfermedad familiar.")
37
INSUFICIENCIA ADRENAL
CAUSAS DE INSUFICIENCIA SUPRARRENAL ETIOLOGÍA (OCURRENCIA %): 1. Insuficiencia adrenal primaria Autoinmune (70%) Tuberculosis (20%) Otras (10%) SIDA Hemocromatosis Infecciones por hongos Adrenoleucodistrofia Hemorragia adrenal Neoplasia metastásica Amiloidosis No respuesta congénita a la ACTH Sarcoidosis Hiperplasia adrenal congénita. 2. Insuficiencia adrenal secundaria Supresión adrenal por administración exógena de glucocorticoides o ACTH Secundario al tratamiento del Síndrome de Cushing Lesiones hipotalámicas o pituitarias.
: 1. Insuficiencia adrenal primaria. Autoinmune (70%) Tuberculosis (20%) Otras (10%) SIDA Hemocromatosis. Infecciones por hongos Adrenoleucodistrofia. Hemorragia adrenal Neoplasia metastásica. Amiloidosis No respuesta congénita a la ACTH. Sarcoidosis. Hiperplasia adrenal congénita. 2. Insuficiencia adrenal secundaria. Supresión adrenal por administración exógena de glucocorticoides o ACTH. Secundario al tratamiento del Síndrome de Cushing. Lesiones hipotalámicas o pituitarias.")
38
INSUFICIENCIA ADRENAL
Las dos causas más comunes de insuficiencia adrenal son la adrenalitis autoinmune y la tuberculosis. En los países en vías de desarrollo, la TB puede ser más prevalente que la adrenalitis autoinmune. La Enfermedad de Addison autoinmune puede ser parte del Síndrome de deficiencia poliendocrina tipo I y II. La tipo I es una enfermedad de la niñez y se caracteriza por insuficiencia adrenal, hipoparatiroidismo, y candidiasis mucocutánea, también puede incluir hipogonadismo, anemia perniciosa, hepatitis crónica activa y alopecia. Tipo II (Síndrome de Schmidt) es una enfermedad de adultos jóvenes, se caracteriza por insuficiencia adrenal, enfermedad autoinmune de la tiroides, y DM tipo 1.
 es una enfermedad de adultos jóvenes, se caracteriza por insuficiencia adrenal, enfermedad autoinmune de la tiroides, y DM tipo 1.")
Presentaciones similares
















 que más se presenta durante la edad adulta, sin embargo también lo hace.>")






