Descargar la presentación
La descarga está en progreso. Por favor, espere

1
ABORDAJE DE LAS MIOPATÍAS
DR. RAFAEL VERA URQUIZA PROFESOR TITULAR: Dr. Enrique Díaz Greene PROFESOR ADJUNTO: Dr. Federico Rodríguez Weber SUPERVISÓ: Dra. Micaela Martínez

2
También conocidas como enfermedad del músculo esquelético.
DEFINICIÓN También conocidas como enfermedad del músculo esquelético. Son trastornos de la estructura primaria o alteración de la función del músculo Goldman: Cecil Medicine, 23rd ed.

3
No es miopatía: Alteraciones del SNC
Enfermedad de la motoneurona inferior Neuropatías periféricas Alteraciones de la placa neuromuscular que generen debilidad muscular Goldman: Cecil Medicine, 23rd ed.

5
Enfrentamiento general a un paciente miopático

6
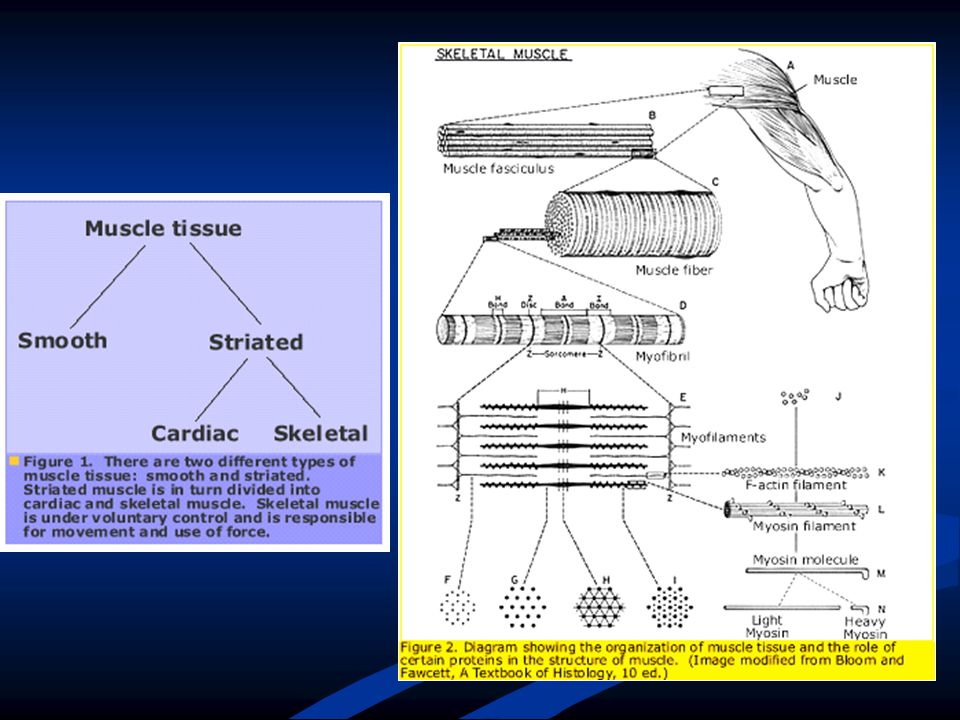
RECORDANDO … Músculo, es el componente fundamental del aparato locomotor y reservorio energético. Cada músculo: miles de fibras musculares. Cada fibra: célula de mm a varios cm (largo), 10 a 100 micras de diámetro. Sarcoplasma. Miofibrillas, retículo sarcoplásmico. Mitocondrias. Ribosomas. Cada fibra recibe un axón. Unidad motora, grupo de fibras musculares con una inervación en común.
, 10 a 100 micras de diámetro. Sarcoplasma. Miofibrillas, retículo sarcoplásmico. Mitocondrias. Ribosomas. Cada fibra recibe un axón. Unidad motora, grupo de fibras musculares con una inervación en común.")
8
Evaluación clínica. Anamnesis. Examen físico. Exámenes:
Edad de inicio. Evolución. Antecedentes familiares. Examen físico. Ver contracción, relajación y reposo. Fatigabilidad. Relación con temperatura. Exámenes: Enzimas musculares. Electromiografía. Biopsia muscular.

9
Patrón de déficits Parálisis ocular. Parálisis facial bilateral.
Ptosis, diplopia, estrabismo (no pupila). Miastenia gravis, POE, DM Oculofaríngea. Parálisis facial bilateral. Miastenia gravis, distrofias y miopatías congénitas. Parálisis bulbar. Miastenia gravis. Debilidad cervical. Polimiositis, miastenia gravis y nemalínica.
. Miastenia gravis, POE, DM Oculofaríngea. Parálisis facial bilateral. Miastenia gravis, distrofias y miopatías congénitas. Parálisis bulbar. Miastenia gravis. Debilidad cervical. Polimiositis, miastenia gravis y nemalínica.")
10
Patrón de déficits Parálisis musc respiratorios. Debilidad proximal.
Maltasa ácida, polimiositis, miastenia gravis. Debilidad proximal. Miositis inflamatorias y DMP. Debilidad distal. Raro. Avanzadas distrofias. Debilidad en musc aislada. No miopático.

11
EMG En reposo. Silencio eléctrico, salud de masa muscular. Si existe contracción es patológico. Fibrilación (denervatorio) y onda aguda positiva. En esfuerzo, potenciales de unidad motora (PUM), voltaje (200 microV a 2 mV), duración (5 a 18 mseg) y número de fases (bi o trifásicos). Miopatía: Actividad espontánea en reposo. Baja amplitud, voltaje
, voltaje (200 microV a 2 mV), duración (5 a 18 mseg) y número de fases (bi o trifásicos). Miopatía: Actividad espontánea en reposo. Baja amplitud, voltaje.")
12
Síntoma cardinal miopatías:
Debilidad muscular. Síntoma cardinal en enfermedades de placa neuromuscular: Fatigabilidad. Diferencia con alt neurogénica: Alt sensibilidad. Topografía déficit motor. Reflejos osteotendíneos. Fasciculaciones. Reflejo ideomuscular disminuido.

13
Goldman: Cecil Medicine, 23rd ed.

14
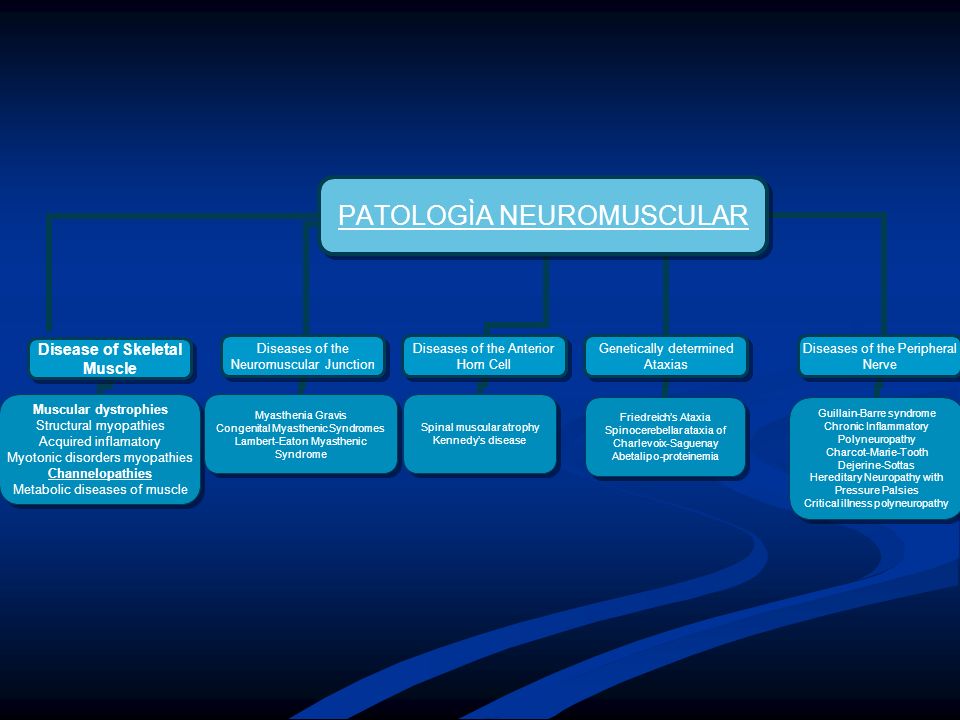
Clasificación de miopatías
Distrofia muscular progresiva. Miopatías inflamatorias. Miopatías metabólicas. Miopatías endocrinas. Miopatías tóxicas. Miopatías congénitas. Miotonías. Miopatía y sd miasténicos.

15
Goldman: Cecil Medicine, 23rd ed.

16
Miopatías congénitas

17
INTRODUCCION M.C. Son un grupo de enfermedades hereditarias que se caracterizan por su comienzo congénito, curso generalmente benigno y presencia de rasgos morfológicos característicos.

19
Clasificación ( modificada )
1- Miopatías con alteración en la maduración o desarrollo muscular: a) Miopatía miotubular asociada al cromosoma X b) Desproporción congénita de tipos de fibras (21%) 2- Miopatías con anormalidades nucleares: a) Miopatías centronucleares (14%) 3- Miopatías con alteración de las proteínas miofibrilares y citoesqueléticas: a) Miopatías concores : - Enfermedad con concores centrales(16%) - Enfermedad con múltiples minicores (10%) b) Miopatía nemalínica. (20%) 180 casos de Fardeau y Tome
 Miopatía miotubular asociada al cromosoma X. b) Desproporción congénita de tipos de fibras (21%) 2- Miopatías con anormalidades nucleares: a) Miopatías centronucleares (14%) 3- Miopatías con alteración de las proteínas miofibrilares y citoesqueléticas: a) Miopatías concores : - Enfermedad con concores centrales(16%) - Enfermedad con múltiples minicores (10%) b) Miopatía nemalínica. (20%) 180 casos de Fardeau y Tome.")
20
Características clínicas
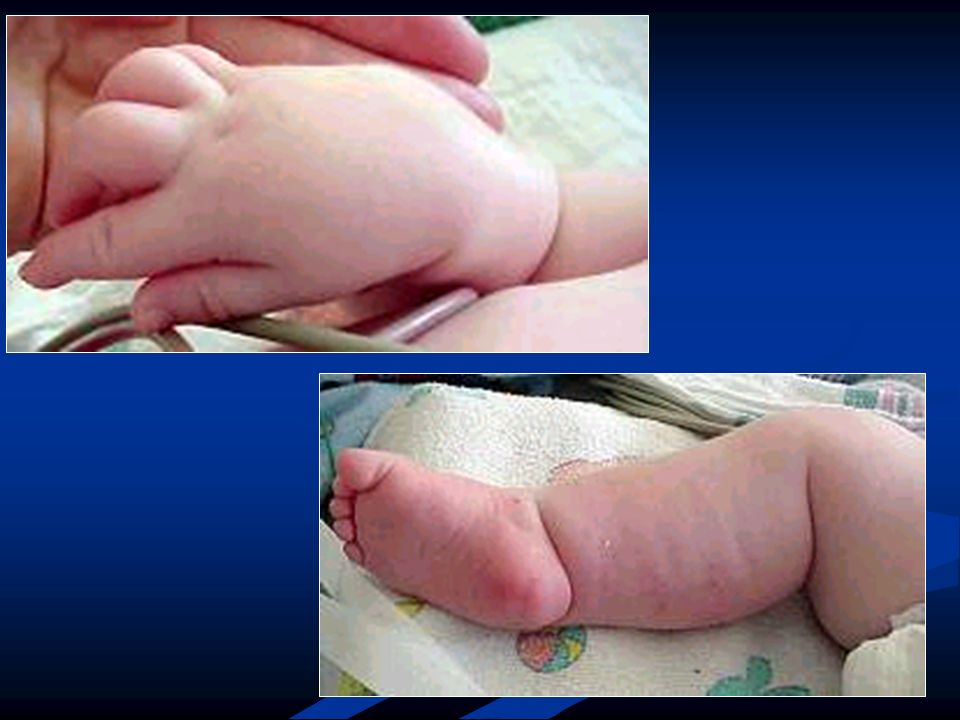
Comparten muchas características clínicas y patológicas, con severidad variable. Inicio precoz. Casos severos: disminución o ausencia de movimientos fetales y partos complicados, seguidos de hipotonía y respiración poco efectiva. Incapacidad para alimentarse.

21
Más comunmente, el primer año presentan hipotonía, debilidad, infecciones respiratorias frecuentes.
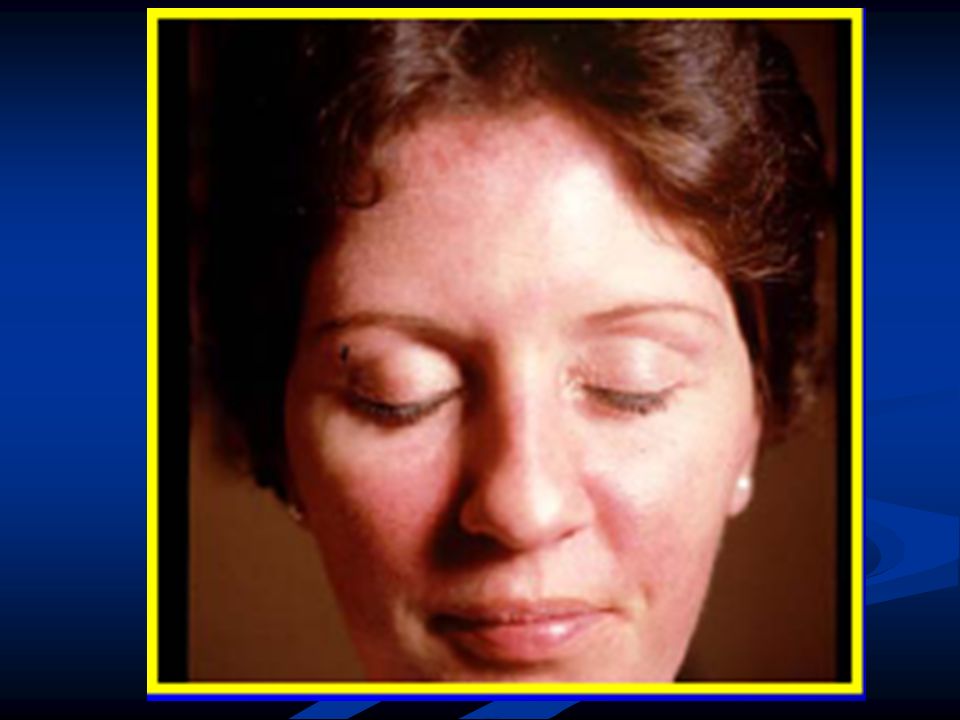
Patrón difuso, predominio proximal. Niños pequeños y delgados. Paresia facial, voz nasal. Raro disfagia. Ptosis y oftalmoparesia puede ocurrir.

22
Laboratorio CK suele ser normal o solo ligeramente elevado.
EFS usualmente normal, a veces un patrón miopático o irritabilidad. No hay evidencia de defecto en unión neuromuscular. A veces la Bp muestra carácter distrófico como aumento de tejido conectivo endomisial. Suele ser necesario más de una biopsia. Inmunohistoquímica suele no ser necesaria para el diagnóstico. Sí ayudaría en la nemalínica con Ac contra alfa- actinina.

23
Miopatía nemalínica La más común de las miopatías congénitas.
Se caracteriza por presencia de bastoncillos o cuerpos nemalínicos en las fibras musculares, generalmente en el sarcoplasma, aunque también pueden aparecer en el núcleo. Patogenia desconocida.

24
Nemaline rod myopathy, Gomori trichrome (GT) stain
Nemaline rod myopathy, Gomori trichrome (GT) stain. Dark blue structures are seen only with this stain. They contain Z disk material, including alpha-actinin and tropomyosin **Bastones también se ven en miopatías VIH, y algunas por drogas.
 stain. Dark blue structures are seen only with this stain. They contain Z disk material, including alpha-actinin and tropomyosin. **Bastones también se ven en miopatías VIH, y algunas por drogas.")
25
MULTIPLES BASTONCILLOS EN MIOPATIA NEMALINICA
Rojo: con Tricrómico Gomori en corte por congelación. Azul con azul toluidina

26
Acúmulos de alfa actinina.

27
En resumen Nemalínica:
Formas precoces: hipotonías congénitas. Debilidad muscular proximal o generalizada, afectación facial.Artrogriposis ocasional. Rostro alargado, inexpresivo. Labio superior en V invertida, paladar ojival. Musc. Extraoculares: no afectados. MFM torácicas, hiperlordosis, espina rígida. Puede presentar pie caído. Fallecen de infecciones respiratorias. Curso lentamente progresivo, la > parte lleva vida activa.

30
Miopatías Inflamatorias

31
Generalidades Grupo heterogeneo de desórdenes caracterizados por inflamación del músculo esquelético, con daño de fibra y debilidad clínica. Existen dos categorías de miopatías. Idiopática. Infecciosa.

32
MIOPATIAS INFLAMATORIAS
Dermatomiositis Polimiositis Miositis por cuerpos de inclusion

34
DIAGNÓSTICO / CLASIFICACIÓN
1. Debilidad muscular (simétrica/proximal) 2. Incremento sérico de un componente muscular intracelular como enzimas (CPK) o mioglobina 3. EMG con anormlidades 4. Inflamación en la biopsia de músculo (1) muscle weakness (usually symmetric and proximal, sparing the muscles innervated by the cranial nerves); (2) an increase in the serum concentration of certain intracellular muscle components, such as enzymes (eg, creatinine kinase) or myoglobin; (3) an electromyogram showing abnormalities; and (4) inflammation seen on muscle biopsy. Classification criteria [3,4] suggest that a diagnosis of inflammatory myopathy is definite if all four of these elements are present, probable when three are present, and possible if two are present. CRITERIOS DE CLASIFICACIÓN: Diagnóstico = 4 elementos Probable = 3 elementos Posible = 2 elementos N Engl J Med 1975; 292(7):344– 7
 2. Incremento sérico de un componente muscular intracelular como enzimas (CPK) o mioglobina. 3. EMG con anormlidades. 4. Inflamación en la biopsia de músculo. (1) muscle weakness (usually symmetric and proximal, sparing the muscles innervated by the cranial nerves); (2) an increase in the serum concentration of certain intracellular muscle components, such as enzymes (eg, creatinine kinase) or myoglobin; (3) an electromyogram showing abnormalities; and (4) inflammation. seen on muscle biopsy. Classification criteria [3,4] suggest that a diagnosis of inflammatory myopathy is definite if all four of these elements are present, probable when three are present, and possible if two are present. CRITERIOS DE CLASIFICACIÓN: Diagnóstico = 4 elementos. Probable = 3 elementos. Posible = 2 elementos. N Engl J Med 1975; 292(7):344– 7.")
35
Dermatomiositis. Se puede presentar a cualquier edad. Incluso infancia. Mujeres se afectan más que hombres. 50 – 60 años. Patogénesis. Es un desorden microangiopático mediado humoralmente. Depósitos vasculares de IgM, C evento inmunológico primario es la generación de Ac contra antígenos de la pared de vasos intramusculares evoluciona a daño isquémico de la fibra muscular.

36
Clínica. El cuadro evoluciona de semanas a pocos meses. Aunque puede presentarse en forma aguda (días) o lentamente progresivo (años). Motor: La debilidad puede ser precedida meses antes de: Fatiga. Dolor y contractura muscular, disminución de la actividad y sd febril. Debilidad: Flexores de cuello. Cintura escapular y pélvica.

37
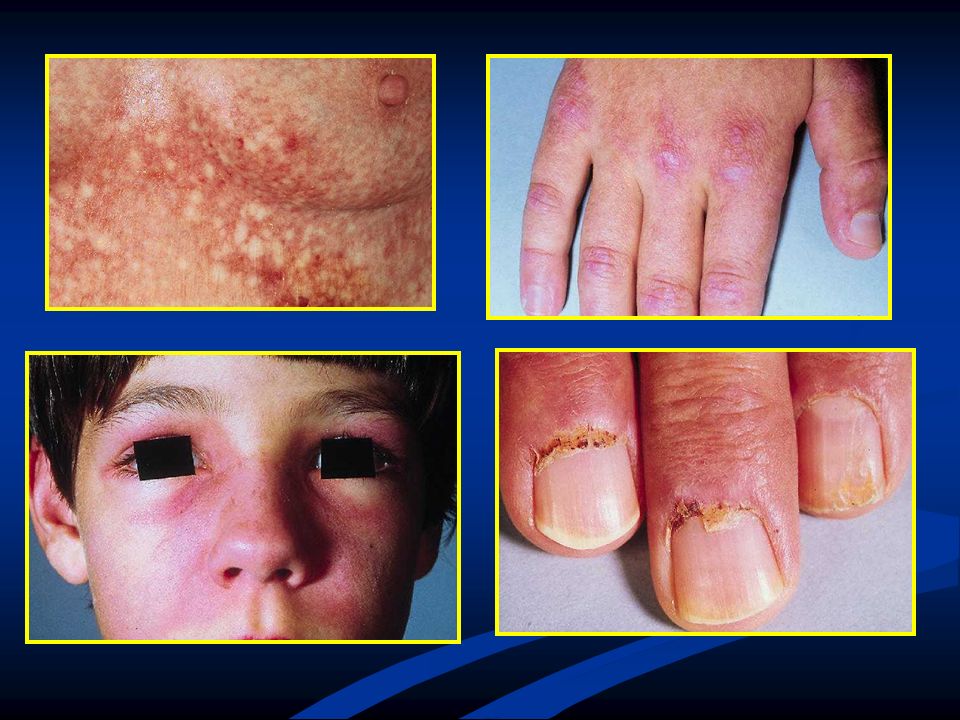
Es mayor la debilidad proximal, aunque tb se puede haber distal.
Quejas: Debilidad en levantarse del piso, de una silla, subir escaleras y llevar EESS sobre su cabeza. Es mayor la debilidad proximal, aunque tb se puede haber distal. 33% presenta disfagia. Raro, disartria, amasia. Rash: Precede a lo motor. Afecta primero a dedos y periorbitario. Decoloración púrpura de párpados (rash heliotropo), con edema periorbitario, mejillas, frente.
, con edema periorbitario, mejillas, frente.")
38
Estas lesiones son dolorosas.
Compromiso de manos: Regiones periungueales con eritema escamoso. Lesiones papular, eritematosas y escamosas sobre nudillos (signo de Gottren). Eritema macular cara, cuello, cara anterior de tórax o sobre hombros y espalda (signo del chal). En niños aparece en 30 a 50% calcificaciones subcutáneas, en adultos no. Estas lesiones son dolorosas. Variante que desarrolla rash pero no debilidad: dermatomiositis amiopática.
. Eritema macular cara, cuello, cara anterior de tórax o sobre hombros y espalda (signo del chal). En niños aparece en 30 a 50% calcificaciones subcutáneas, en adultos no. Estas lesiones son dolorosas. Variante que desarrolla rash pero no debilidad: dermatomiositis amiopática.")
42
Laboratorio. Evaluación incluye:
Pruebas musculares. Electrocardiograma. Biopsia de músculo. Creatinin cinasa, marcador más seguro, sensible y específico de destrucción muscular, elevado en más del 90% de los pctes. Niveles sobre 50 veces el valor normal. Los niveles se correlacionan con la gravedad de la debilidad.

43
Aldolasa. Mioglobina. Lactato deshidrogenasa Elevados, pero Aspartato aminotransferasa No aportan mayor Alanina aminotransferasa información. VHS elevado levemente, no correlacionado con gravedad. ANA + en 25 a 50%. Ac miositis específicos están asociados a HLA (limitados en clínica). Ac Jo – 1 + en pctes con enfermedad intersticial pulmonar.
. Ac Jo – 1 + en pctes con enfermedad intersticial pulmonar.")
44
Electromiografía Útil en la demostración de la naturaleza miopática del desorden. Ayuda en la determinación de que músculo biopsiar en casos leves. Aumento de la actividad espontánea. Potenciales de fibrilación. Positive sharp waves. Ocasionalmente descargas pseudomiotónicas o complejos repetitivos.

45
Tratamiento. Corticoesteroides.
Controversia en vía de dosis, régimen de dosis, duración de terapia y parámetros de monitoreo. Trials no controlados y retrospectivos: Prednisona reduce mortalidad y mejora función. Cuadros severos: Solumedrol IV 20 a 30 mg/Kg/al día por 3 a 5 días. VO 1,5 mg/Kg/día como dosis única en la mañana. Luego de 2 a 3 semanas de terapia VO, cambiar a régimen a dias alternativo. NO CK como determinante monitoreo.

46
Pcte debe ser controlado 1 vez al mes.
Cuando las fuerzas han retornado o se produjo un plateau (4 a 6to mes), se puede iniciar tapering bajando 5 mg cada 2 a 4 semanas. Si hay remitencia en este período dar 1,5 mg/Kg diario. Ig IV: Efecto benéfico en trials doble ciegos y estudios controlados. 2g/Kg entre 2 a 5 días. Repetido cada 2 a 6 semanas por 3 meses. 50% de síntomas flu – lika.
, se puede iniciar tapering bajando 5 mg cada 2 a 4 semanas. Si hay remitencia en este período dar 1,5 mg/Kg diario. Ig IV: Efecto benéfico en trials doble ciegos y estudios controlados. 2g/Kg entre 2 a 5 días. Repetido cada 2 a 6 semanas por 3 meses. 50% de síntomas flu – lika.")
47
Polimiositis. Entre los 50 a 60 años. Raro en niños y cuando ocurre es dentro de un “sd de overlap”. Clínicamente se presenta similar a dermatomiositis, evolución de semanas a meses, con debilidad de flexores de cuello y proximal simétrico. Puede estar asociado a dolor. 30% disfagia. 10% enf intersticial pulmonar (Ac anti Jo – 1). Poliartritis en 45%. Músculo cardíaco puede estar comprometido.
. Poliartritis en 45%. Músculo cardíaco puede estar comprometido.")
48
Laboratorio. CK sérica elevada 50 veces el valor normal. ANA + en 30%.
Ac miositis específicos +. Ac anti Jo – 1 en 20%. EMG: similar a dermatomiositis. Con patrón miopático de actividad espontánea aumentada, unidades motoras polifásicas pequeñas y reclutamiento temprano.

49
Tratamiento. No difiere de dermatomiositis. Agente ppal, corticoides.
Papel de Ig IV es menos conocido que en dermatomiositis. En general la respuesta es menor que en dermatomiositis.

51
Distrofias musculares.

52
Las distrofias musculares son miopatìas genéticamente determinadas, usualmente causadas por un disturbio en la síntesis de una proteína estructural específica. También las podríamos definir como entidades que afectan principalmente al músculo estriado y que tienen en comùn un patròn distròfico de necrosis-regeneraciòn caracterìstico en la biopsia muscular.

53
Componentes del citoesqueleto de la fibra muscular y del sarcolema:
La mayor parte de las distrofias tienen como causa anormalidades que involucran a las llamadas proteìnas estructurales. Componentes del citoesqueleto de la fibra muscular y del sarcolema: Complejo de Glicoproteìnas asociadas a la distrofina.

54
DISTROFIAS MUSCULARES PROGRESIVAS
Type Onset Age (years) Clinical Features Other organ systems involved Duchenne Before 5 Progressive weakness of girdle muscles. unable to walk after age 12 progressive kyphoscoliosis Respiratory failure in 2dor 3d decade. Cardiomyopathy Mental impairment Becker 5-25yr early childhood to adult Progressive weakness of girdle muscles 2. able to walk after age 15. 3. respiratory failure may develop by 4th grade Emery-Dreifuss Childhood to adult Elbow contractures, humeral and perineal weakness Limb-Girdle Slow progressive weakness of shoulder and hip girdle muscles
 Clinical Features. Other organ systems involved. Duchenne. Before 5. Progressive weakness of girdle muscles. unable to walk after age 12. progressive kyphoscoliosis. Respiratory failure in 2dor 3d decade. Cardiomyopathy. Mental impairment. Becker. 5-25yr. early childhood to adult. Progressive weakness of girdle muscles. 2. able to walk after age respiratory failure may develop by 4th grade. Emery-Dreifuss. Childhood to adult. Elbow contractures, humeral and perineal weakness. Limb-Girdle. Slow progressive weakness of shoulder and hip girdle muscles.")
55
Other organ systems involved
DISTROFIAS MUSCULARES PROGRESIVAS Type Onset Age (years) Clinical Features Other organ systems involved Congenital At birth or within 1st few months .Hypotonia, contractures, delayed milestones Progression to respiratory failure in some; CNS and Eye abnormalities Facioscapulohumeral Before age 20 Slowly progressive weakness of face, shoulder girdle, and foot dorsiflexion Deafness Coat’s (eye) disease Oculopharyngeal 5th to 6th decade Slowly progressive weakness of extraocular, pharyngeal, and limb muscles ______ Myotonic Usually 2nd decade May be infancy if mother affected Cardiac conduction defects Mental impairment Cataracts Frontal baldness Gonadal atrophy
 Clinical Features. Other organ systems involved. Congenital. At birth or within 1st few months. .Hypotonia, contractures, delayed milestones. Progression to respiratory failure in some; CNS and. Eye abnormalities. Facioscapulohumeral. Before age 20. Slowly progressive weakness of face, shoulder girdle, and foot dorsiflexion. Deafness. Coat’s (eye) disease. Oculopharyngeal. 5th to 6th decade. Slowly progressive weakness of extraocular, pharyngeal, and limb muscles. ______. Myotonic. Usually 2nd decade. May be infancy if mother affected. Cardiac conduction defects. Mental impairment. Cataracts. Frontal baldness. Gonadal atrophy.")
56
GUILLERMO DUCHENNE

57
DISTROFIA DE DUCHENNE Y BECKER
Enfermedad de herencia recesiva ligada al cromosoma X. Dicho gen codifica la producciòn de distrofina. En la distrofia de Duchenne hay total carencia de distrofina. Frecuencia: de 1:3.500.

58
DISTROFIA DE DUCHENNE Y BECKER
Comienza entre los 2 y 4 años con retraso motor (40%), marcha anormal (30%), transtorno del lenguaje y el habla (8%). Excepcionalmente el inicio de los sìntomas es muy precoz, expresado por hipotonìa desde la lactancia temprana. Los signos caracterìsticos son: Debilidad de musculatura escàpulo-pèlvica, hipertrofia o seudohipertrofia gemelar, debilidad de los flexores del cuello.
, marcha anormal (30%), transtorno del lenguaje y el habla (8%). Excepcionalmente el inicio de los sìntomas es muy precoz, expresado por hipotonìa desde la lactancia temprana. Los signos caracterìsticos son: Debilidad de musculatura escàpulo-pèlvica, hipertrofia o seudohipertrofia gemelar, debilidad de los flexores del cuello.")
59
DISTROFIA DE DUCHENNE Y BECKER
CI lìmite y ràpida progresiòn. Apariciòn posterior de retracciòn aquiliana, escoliosis y pèrdida de la ambulaciòn antes de los trece años. Signo de Gowers o maniobra de pararse trepando sobre si mismo es positivo. La expectativa de vida no sobrepasa la mitad de la tercera dècada. La muerte se debe a fallo respiratorio agudo o miocardiopatìa dilatada refractaria a tratamiento.

60
MANIOBRA DE GOWERS

61
DISTROFIA DE DUCHENNE Y BECKER
Estudios: CK elevada: Hasta mu/ml. Estudios de conducciòn nerviosa: Normales. EMG de aguja en reposo: Presencia de fibrilaciòn, ondas positivas y descargas de alta frecuencia. Patròn voluntario està compuesto por unidades motoras en general polifàsicas, breves y de baja amplitud.

62
DISTROFIA DE DUCHENNE Y BECKER
Biopsia muscular: Muestra un patròn distròfico con intensa fibrosis endomisial, fibras hipercontraìdas, diferentes grados de necrosis. Tècnicas de inmunohistoquìmica y de Western Blot: Se demuestra una ausencia total de distrofina.

63
DISTROFIA DUCHENNE MICROSCOPÌA

64
DISTROFIA DE DUCHENNE Y BECKER
DISTROFIA DE BECKER: Frecuencia: 1: varones. Sintomatologìa: Se inicia generalmente a los 5-15 años de edad, aunque los casos màs leves son de presentaciòn màs tardìa. Patròn de debilidad muscular similar a la de Duchenne. Con frecuencia existe intolerancia al ejercicio y mioglobinuria, y en ocasiones calambres.

65
DISTROFIA DE DUCHENNE Y BECKER
La clìnica es menos grave que en la forma Duchenne y los pacientes pierden la deambulaciòn unos 16 años despuès del comienzo de la enfermedad o incluso màs tardiamente. No es infrecuente la afectaciòn cardiaca, generalmente en forma de miocardiopatìa dilatada. EMG: Hallazgos menos acentuados que en la forma Duchenne.

67
DISTROFIA OCULO-FARINGEA

68
DISTROFIA FACIOESCÀPULOHUMERAL

69
Miopatías mitocondriales

72
Abordaje diagnóstico de enfermedades mitocondriales

73
Determinación del acido láctico
Enzimas musculares EMG Biopsia muscular Microscopia electrónica Neuroimágenes Estudio molecular

74
Enzimas musculares Suelen ser normales o discretamente elevadas.

75
Biopsia muscular La fibras rojo rasgadas son caracteristicas
(tincion de tricromico Gomori) y expresan un cambio morfologico secundario a una fosforilacion oxidativa defectuosa. Son fibras que se forman al cortar el músculo congelado se tiñen rojo en la periferia por acumulo de mitocondrias. Aparecen en mutaciones y deleciones del ADNmt que codifica RNAt no se ven en los que codifican proteinas estructurales (NARP y MILS)
 y expresan un cambio morfologico secundario a una fosforilacion oxidativa defectuosa. Son fibras que se forman al cortar el músculo congelado se tiñen rojo en la periferia por acumulo de mitocondrias. Aparecen en mutaciones y deleciones del ADNmt que codifica RNAt no se ven en los que codifican proteinas estructurales (NARP y MILS)")
77
Oftalmoplejia externa progresiva.
Sd de Kearns – Sayre. Encefalomiopatia mitocondrial con acidosis láctica y episodios de stroke-like (MELAS). Sindrome de Leigh de herencia materna (MILS). Encefalomiopatia neurogastrointestinal mitocondrial (MNGIE). Neuropatia ataxica sensitiva con disartria y oftalmoparesia (SANDO)
. Sindrome de Leigh de herencia materna (MILS). Encefalomiopatia neurogastrointestinal mitocondrial (MNGIE). Neuropatia ataxica sensitiva con disartria y oftalmoparesia (SANDO)")
Presentaciones similares