Descargar la presentación
La descarga está en progreso. Por favor, espere

1
DIA INTERNACIONAL DE LA ENFERMERA
12 de Mayo de 2011 DIA INTERNACIONAL DE LA ENFERMERA

2
ENSAYO CLÍNICO: LA PRÁCTICA Y SUS PROBLEMAS

3
¿Qué es un comité de ética en investigación clínica?
Objetivos de hoy ¿Qué es un comité de ética en investigación clínica? Un ensayo clínico: Ensayos clínicos en oncología

4
¿Qué es un comité de ética en investigación clínica?

5
INVESTIGACIÓN Y PROGRESO

6
INVESTIGACIÓN Y PROGRESO
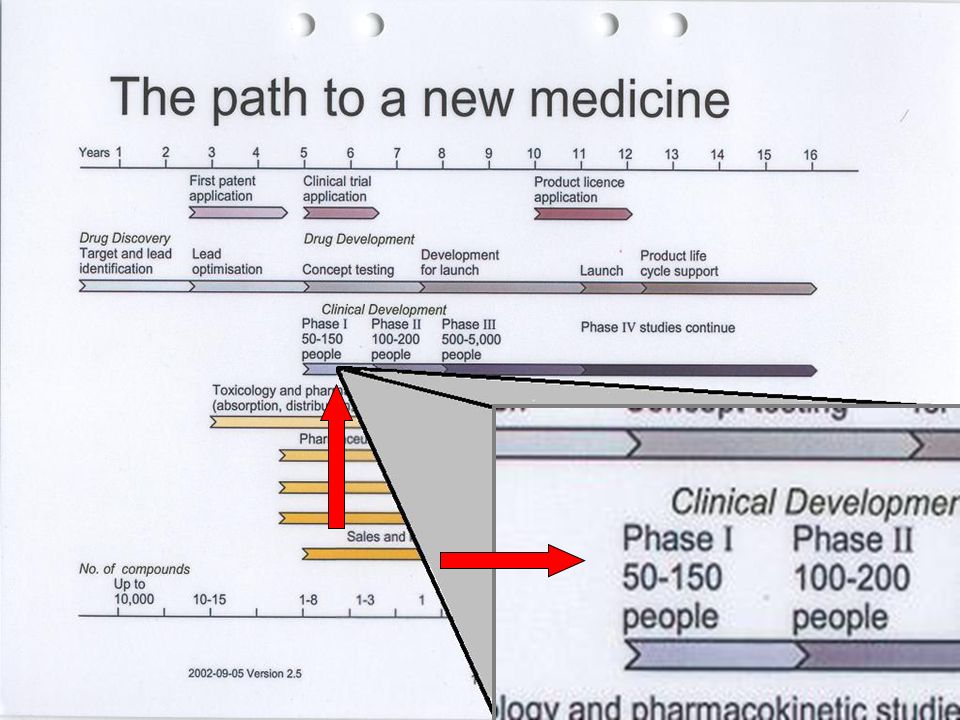
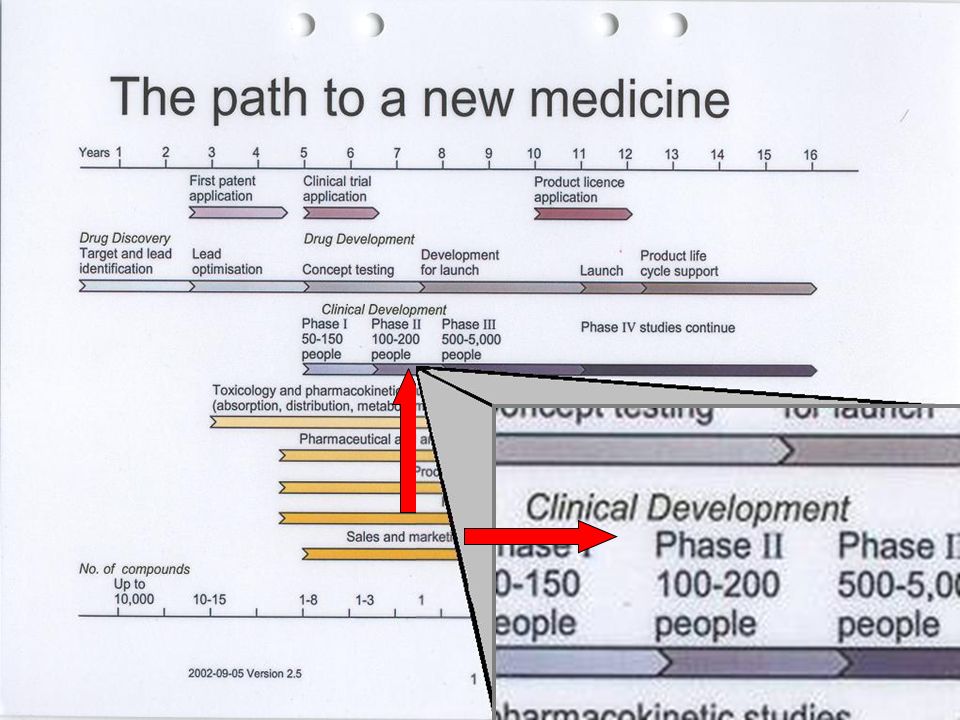
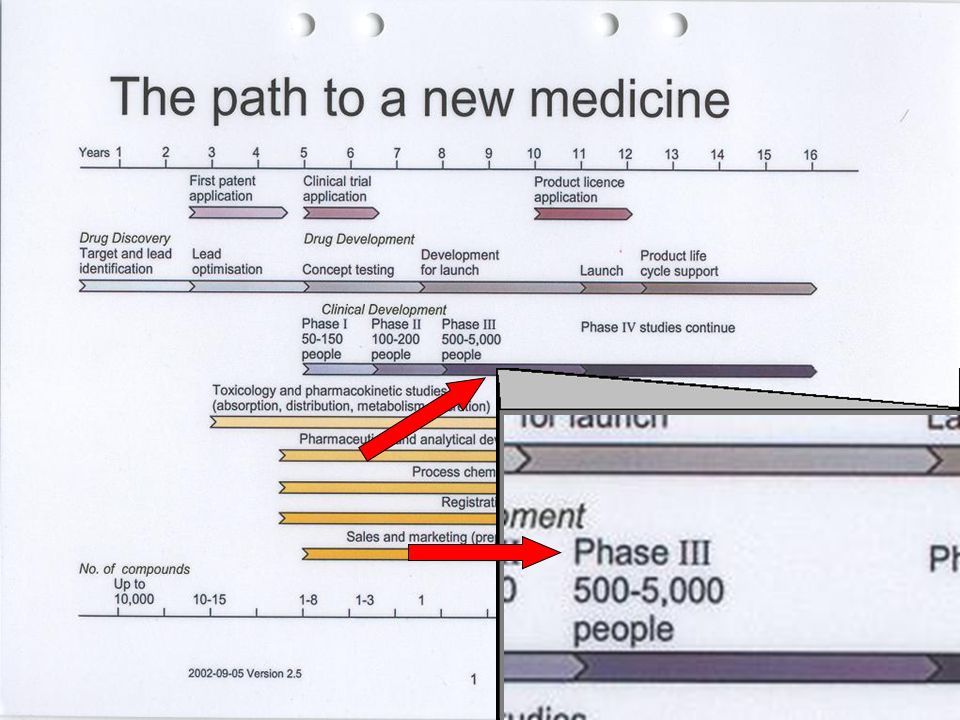
Industria Farmacéutica Investiga Desarrolla Fabrica y Comercializa Invierte: Medios Humanos Tecnológicos Económicos 1/ moléculas 8 a 10 años Coste 500 $ Los medicamentos son investigados, desarrollados, fabricados y comercializados por la Industria Farmacéutica, que invierte en estas actividades un esfuerzo considerable en medios humanos, tecnológicos y económicos con un lícito ánimo de lucro. En la actualidad, sólo una de cada diez mil moléculas investigadas llega a comercializarse como medicamento, después de un plazo de 8 a 10 años de investigación y desarrollo con un coste económico de alrededor de 500 millones de dólares. Ánimo de lucro investiga, desarrolla, fabrica y comercializa. invierte medios humanos, tecnológicos y económicos con ánimo de lucro. Sólo una de cada diez mil moléculas investigadas llega a comercializarse como medicamento, después de un plazo de 8 a 10 años de investigación y desarrollo con un coste económico de alrededor de 500 millones de dólares. INVESTIGACIÓN Y PROGRESO El plan de desarrollo clínico La legislación española normas de Buena Práctica Clínica (BPC).
.")
7
Nueva molécula para uso humano
Plan de desarrollo clínico: Exhaustivo conocimiento del producto Demostrar seguridad y eficacia en las patologías indicada. Agencia Española del Medicamento: solicitar autorización ley 14/1986 General de Sanidad para la circulación y uso de los medicamentos y productos sanitarios. ¿cuál es el procedimiento que debe seguir para que ésta llegue a convertirse en medicamento de uso humano? En primer lugar deberá realizar un completo plan de desarrollo clínico con el fin de obtener un exhaustivo conocimiento del producto y demostrar su seguridad y eficacia en las patologías para las que está indicada. Posteriormente deberá aportar la información obtenida a la agencia reguladora correspondiente (la Agencia Española del Medicamento) solicitando su autorización, de acuerdo con la ley 14/1986 General de Sanidad(2), la cual dispone que para la circulación y uso de los medicamentos y productos sanitarios se requiere autorización previa
 solicitando su autorización, de acuerdo con la ley. 14/1986 General de Sanidad(2), la cual dispone que para la circulación y uso de los medicamentos y. productos sanitarios se requiere autorización previa.")
8
Manual del instructor para Comités de Asesoría Comunitaria
Module 2: Introduction to Clinical Trials El plan de desarrollo clínico: Realización de ensayos clínicos con el producto a investigar cuatro fases

9
Fases de los ensayos clínicos
Manual del instructor para Comités de Asesoría Comunitaria Fases de los ensayos clínicos Module 2: Introduction to Clinical Trials Fase I: entre 15 y 30 personas Fase II Menos de 100 personas Fase III: de 100 a varios miles de personas Los ensayos clínicos que prueban medicamentos o tratamientos nuevos se realizan en fases. El ensayo empieza con ensayos clínicos Fase I, con muy pocos participantes, para probar la seguridad del tratamiento. Si no hay problemas graves, los ensayos continúan con la fase II y luego en la fase III A veces también se realiza una Fase IV. Las primeras fases solamente estudian la inocuidad (es decir, que el medicamento no haga daño). Para determinar la eficacia de un tratamiento, en general, los investigadores deben estudiarlo en un mayor número de personas en los ensayos Fase III. Es posible que las compañías farmacéuticas (y, a veces otros) continúen recopilando información incluso después de que un medicamento se haya aprobado. Esto les ayuda a proporcionar información acerca de los beneficios o problemas a largo plazo. Este tipo de investigación es un estudio "Fase IV" o de "poscomercialización". En las próximas diapositivas veremos más detalles de cada una de estas fases. Fase IV: >Poscomercialización. Las dimensiones del estudio varían de acuerdo con las prácticas de prescripción.
. Para determinar la eficacia de un tratamiento, en general, los investigadores deben estudiarlo en un mayor número de personas en los ensayos Fase III. Es posible que las compañías farmacéuticas (y, a veces otros) continúen recopilando información incluso después de que un medicamento se haya aprobado. Esto les ayuda a proporcionar información acerca de los beneficios o problemas a largo plazo. Este tipo de investigación es un estudio Fase IV o de poscomercialización . En las próximas diapositivas veremos más detalles de cada una de estas fases. Fase IV: >Poscomercialización. Las dimensiones del estudio varían de acuerdo con las prácticas de prescripción.")
10
Manual del instructor para Comités de Asesoría Comunitaria
Module 2: Introduction to Clinical Trials Fase I: Inocuidad Meta: determinar si un medicamento es inocuo en un pequeño grupo de voluntarios sanos. Si un medicamento nuevo funciona bien en las pruebas de laboratorio y es inocuo cuando se lo prueba en animales, el paso siguiente es realizar un estudio Fase I para determinar si es seguro para los seres humanos. Los ensayos clínicos Fase I se centran en probar el medicamento en aproximadamente 10 a 30 voluntarios. Prueban el medicamento en sujetos sanos porque es menos probable que tengan efectos adversos graves debido a los medicamentos. Por ejemplo, un nuevo medicamento ARV para el VIH se probará primero en el laboratorio, luego en animales y luego en un estudio Fase I en una pequeño grupo de participantes que no tengan VIH y que, en general, sean sanos. En los estudios Fase I, la mayoría de las veces, los participantes reciben el medicamento solamente unos pocos días. Durante este tiempo, los investigadores realizan un seguimiento estrecho de los participantes. Por esta razón, los estudios Fase I suelen realizarse en hospitales.

11
Manual del instructor para Comités de Asesoría Comunitaria
Module 2: Introduction to Clinical Trials Inocuidad y Eficacia Estudio Fase II: Si no hay problemas graves de seguridad en la Fase I, el medicamento se evalúa en un estudio Fase II con más personas. Si los investigadores no encuentran problemas de seguridad en el ensayo clínico Fase I, prueban el medicamento en un ensayo clínco Fase II con un grupo más numeroso de personas (entre 31 y 100). A diferencia de los participantes sanos de los estudios Fase I, los participantes en los estudios Fase II sí tienen la enfermedad en estudio. A diferencia de los estudios Fase I, un nuevo medicamento ARV para el VIH en un estudio Fase II se probará en personas infectadas con el virus. Si se realiza un ensayo clínico paraprevenir en vez de para tratar una enfermedad, es posible que el participanteno tenga la enfermedad. Por ejemplo, en un estudio de un medicamento para evitar la transmisión maternoinfantil del VIH, el objetivo es evitar que el bebé contraiga la enfermedad. Las metas de un estudio Fase II son determinar si el medicamento es inocuo y eficaz y averiguar la cantidad de medicamento que una persona debe tomar para que sea eficaz. Los estudios para determinar la cantidad necesaria de medicamento se llaman estudios de búsqueda de dosis. Los estudios Fase II, a veces, son estudios de búsqueda de dosis. En general, estos estudios requieren muchos análisis de sangre para determinar la cantidad de medicamento en sangre en diferentes momentos. Eso se conoce como pruebas farmacocinéticas o pruebas PK (de pharmacokinetic, en inglés). Este tipo de pruebas, si bien son difíciles, son fundamentales para encontrar la dosis correcta que sea inocua y a la vez eficaz.
. A diferencia de los participantes sanos de los estudios Fase I, los participantes en los estudios Fase II sí tienen la enfermedad en estudio. A diferencia de los estudios Fase I, un nuevo medicamento ARV para el VIH en un estudio Fase II se probará en personas infectadas con el virus. Si se realiza un ensayo clínico paraprevenir en vez de para tratar una enfermedad, es posible que el participanteno tenga la enfermedad. Por ejemplo, en un estudio de un medicamento para evitar la transmisión maternoinfantil del VIH, el objetivo es evitar que el bebé contraiga la enfermedad. Las metas de un estudio Fase II son determinar si el medicamento es inocuo y eficaz y averiguar la cantidad de medicamento que una persona debe tomar para que sea eficaz. Los estudios para determinar la cantidad necesaria de medicamento se llaman estudios de búsqueda de dosis. Los estudios Fase II, a veces, son estudios de búsqueda de dosis. En general, estos estudios requieren muchos análisis de sangre para determinar la cantidad de medicamento en sangre en diferentes momentos. Eso se conoce como pruebas farmacocinéticas o pruebas PK (de pharmacokinetic, en inglés). Este tipo de pruebas, si bien son difíciles, son fundamentales para encontrar la dosis correcta que sea inocua y a la vez eficaz.")
12
Manual del instructor para Comités de Asesoría Comunitaria
Module 2: Introduction to Clinical Trials Fase III: Inocuidad y Eficacia Si los resultados de la Fase II demuestran que el medicamento no tiene problemas importantes de inocuidad y parece ser eficaz, los investigadores realizan un estudio Fase III. Si los resultados del estudio Fase II muestran que el medicamento es inocuo y eficaz, los investigadores llevan a cabo un estudio Fase III. Esta fase incluye un número todavía mayor de pacientes (cientos o miles). En el estudio Fase III, los investigadores continúan evaluando la inocuidad de un medicamento pero se centran cada vez más en cómo actúa (grado de eficacia) para tratar o prevenir la enfermedad.
. En el estudio Fase III, los investigadores continúan evaluando la inocuidad de un medicamento pero se centran cada vez más en cómo actúa (grado de eficacia) para tratar o prevenir la enfermedad.")
13
Manual del instructor para Comités de Asesoría Comunitaria
Module 2: Introduction to Clinical Trials Fase III: Eficacia e Inocuidad Tratamiento experimental Metas Determinar si un medicamento actúa bien(eficacia). Continuar evaluando la inocuidad. en comparación con Tratamiento estándar En los estudios Fase III, los investigadores también evalúan cómo actúa el nuevo medicamento en comparación con un medicamento que ya sepan que es inocuo y eficaz. Este medicamento conocido se suele llamar tratamiento estándar de la enfermedad. Este tipo de estudio se llama ensayo clínico de comparación o ensayo clínico controlado. (Más adelante en esta capacitación hablaremos qué significa "controlado" en la descripción de un estudio). Por ejemplo, un estudio Fase III podría comparar un nuevo medicamento para tratar una infección por el VIH con un medicamento para el virus que ya esté en uso y se sepa que es eficaz para tratar el virus. Comparar los datos provenientes de ambos grupos es una manera mucho más precisa de estudiar un nuevo medicamento que simplemente observar los efectos del nuevo medicamento en un grupo de personas.
. Continuar evaluando la inocuidad. en comparación con. Tratamiento estándar. En los estudios Fase III, los investigadores también evalúan cómo actúa el nuevo medicamento en comparación con un medicamento que ya sepan que es inocuo y eficaz. Este medicamento conocido se suele llamar tratamiento estándar de la enfermedad. Este tipo de estudio se llama ensayo clínico de comparación o ensayo clínico controlado. (Más adelante en esta capacitación hablaremos qué significa controlado en la descripción de un estudio). Por ejemplo, un estudio Fase III podría comparar un nuevo medicamento para tratar una infección por el VIH con un medicamento para el virus que ya esté en uso y se sepa que es eficaz para tratar el virus. Comparar los datos provenientes de ambos grupos es una manera mucho más precisa de estudiar un nuevo medicamento que simplemente observar los efectos del nuevo medicamento en un grupo de personas.")
14
Manual del instructor para Comités de Asesoría Comunitaria
Module 2: Introduction to Clinical Trials Estudio Fase IV Meta: Determinar la inocuidad y la eficacia a largo plazo en un gran número de personas. Los estudiosFase IV se realizan despuésde que se haya aprobado el nuevo medicamento para la venta y de que se lo utilice. A menudo se los llama estudios de poscomercialización porque los medicamentos ya han sido aprobados y pueden recetarse y venderse. En estos ensayos clínicos se obtienen datos solamente de las personas a las que sus médicos clínicos les han recetado el medicamento. No hay grupo control ni de comparación. Estos estudios se realizan principalmente para evaluar los efectos a largo plazo del medicamento. A veces, se pueden ver los problemas o los buenos efectos de un medicamento solamente después de que los participantes hayan tomado el medicamento muchos años. En general, estos estudios los hacen las compañías farmacéuticas que desarrollaron el medicamento y no las redes de ensayos clínicos como IMPAACT. Pregunta para debatir: Si los investigadores ya saben que el medicamento sirve y es inocuo, ¿por qué son tan importantes los estudios Fase IV? ¿Se acuerdan de algún ejemplo de un problema a largo plazo con un medicamento que se haya descubierto despuésde que haya sido aprobado y vendido? Ejemplo: Con el tiempo se descubrió que una clase de medicamentos que se usaba para la artritis aumentaba el riesgo de ataques cardíacos en personas que tomaban uno de esos medicamentos. Esto no se supo en los ensayos clínicos de las fases anteriores. Se hizo evidente una vez que se dispuso de datos de miles de personas durante largo tiempo.

15
Manual del instructor para Comités de Asesoría Comunitaria
DISEÑO Manual del instructor para Comités de Asesoría Comunitaria Module 2: Introduction to Clinical Trials Estudio controlado Grupo experimental Muestra del estudio Resultado A en comparación con Grupo control Ahora vamos a hablar un poco más de lo que se llama el “plan o el diseño del estudio”. Los investigadores deben diseñar un estudio de manera que logren la mejor forma, es decir, la forma más científica de obtener la respuesta más precisa a la pregunta de investigación. La diapositiva muestra el plan para un ensayo clínico controlado que compara dos medicamentos. A la izquierda pueden ver la muestra en estudio, donde se incluye a todos los participantes en el ensayo clínico. A la derecha, pueden ver que se ha asignado a cada uno de los voluntarios de la muestra al grupo experimental (o sea, al que recibe el medicamento nuevo) o al grupo control (el que recibe el medicamento estándar). Al cabo de varias semanas de que el grupo experimental haya tomado el medicamento nuevo y que el grupo control haya tomado el medicamento estándar, los investigadores comparan los resultados de los dos grupos. Y así procurarán averiguar si el nuevo medicamento tiene mayor, igual o menor eficacia que el medicamento estándar. Tan importante como eso es que averigüen si el nuevo medicamento es inocuo, es decir, si no causa daño, si es seguro. B Resultado
 o al grupo control (el que recibe el medicamento estándar). Al cabo de varias semanas de que el grupo experimental haya tomado el medicamento nuevo y que el grupo control haya tomado el medicamento estándar, los investigadores comparan los resultados de los dos grupos. Y así procurarán averiguar si el nuevo medicamento tiene mayor, igual o menor eficacia que el medicamento estándar. Tan importante como eso es que averigüen si el nuevo medicamento es inocuo, es decir, si no causa daño, si es seguro. B. Resultado.")
16
Aleatorización de participantes
Manual del instructor para Comités de Asesoría Comunitaria Module 2: Introduction to Clinical Trials Aleatorización de participantes Grupo Experimental Asignación de participantes a los distintos grupos mediante un proceso que se llama aleatorización. A Muestra del estudio En esta imagen pueden ver que aproximadamente la mitad de la muestra del estudio participará en el grupo experimental (también llamado grupo de tratamiento) y que la otra mitad aproximada participará en el grupo control. El investigador no elige el grupo en que estará cada participante. Tampoco se le permite elegir a los participantes. Una computadora decide quién estará en qué grupo mediante un proceso llamado aleatorización. Este es un ejemplo de cómo funciona la aleatorización. Imagínense que tenemos un gran frasco con una mezcla en igual proporción, 50-50, de cuentas azules y rojas. Cuando un participante se inscribe en un estudio, es como si tomara una cuenta del jarro con los ojos vendados. Ni el participante ni nadie más controla qué cuenta escoge... la elección de la cuenta es aleatoria absolutamente al azar. Si la cuenta roja indicara el grupo experimental y la azul el grupo control, aproximadamente la mitad de la muestra de participantes participaría en cada grupo. Lo que es más, es probable que los grupos también tengan otras características distribuidas equitativamente (por ejemplo, el número de hombres y mujeres). La aleatorización es muy importante porque evita el sesgo (es decir, la parcialidad, el favorecer a un grupo o persona más que a otros). En la red de investigación, no usamos cuentas, sino que una computadora distribuye aleatoriamente a los participantes al azar, sin que ni los investigadores ni los participantes controlen la distribución, Demostración rápida de aleatorización: Para demostrar la aleatorización, coloque sobre la mesa una pila de dos tipos de cuentas, frijoles, porotos, fideos u otro elemento común. El número de elementos en la sobre la mesa debe ser igual al número de participantes en el ejercicio. Pida a los participantes que se acerquen de a uno a la mesa. El participante debe cerrar los ojos, el instructor mezcla un poco la pila, y el participante escoge un elemento. Grupo control
 y que la otra mitad aproximada participará en el grupo control. El investigador no elige el grupo en que estará cada participante. Tampoco se le permite elegir a los participantes. Una computadora decide quién estará en qué grupo mediante un proceso llamado aleatorización. Este es un ejemplo de cómo funciona la aleatorización. Imagínense que tenemos un gran frasco con una mezcla en igual proporción, 50-50, de cuentas azules y rojas. Cuando un participante se inscribe en un estudio, es como si tomara una cuenta del jarro con los ojos vendados. Ni el participante ni nadie más controla qué cuenta escoge... la elección de la cuenta es aleatoria absolutamente al azar. Si la cuenta roja indicara el grupo experimental y la azul el grupo control, aproximadamente la mitad de la muestra de participantes participaría en cada grupo. Lo que es más, es probable que los grupos también tengan otras características distribuidas equitativamente (por ejemplo, el número de hombres y mujeres). La aleatorización es muy importante porque evita el sesgo (es decir, la parcialidad, el favorecer a un grupo o persona más que a otros). En la red de investigación, no usamos cuentas, sino que una computadora distribuye aleatoriamente a los participantes al azar, sin que ni los investigadores ni los participantes controlen la distribución, Demostración rápida de aleatorización: Para demostrar la aleatorización, coloque sobre la mesa una pila de dos tipos de cuentas, frijoles, porotos, fideos u otro elemento común. El número de elementos en la sobre la mesa debe ser igual al número de participantes en el ejercicio. Pida a los participantes que se acerquen de a uno a la mesa. El participante debe cerrar los ojos, el instructor mezcla un poco la pila, y el participante escoge un elemento. Grupo control.")
17
Sin aleatorización = sesgo
Manual del instructor para Comités de Asesoría Comunitaria Module 2: Introduction to Clinical Trials Sin aleatorización = sesgo Grupo experimental Muestra del estudio A Grupo control Esta diapositiva muestra un ejemplo de cómo puede introducirse sesgo si no se aplica aleatorización a un estudio. Imagine que los participantes con mayor carga viral fueran los más interesados en el medicamento experimental porque creyeran que les ayudaría más que el medicamento estándar que se le administrará al grupo control. Por lo tanto, es mucho más probable que los participantes con cargas virales altas (las personas azules) elijan participar en el grupo experimental. Al contrario, los participantes con cargas virales bajas, al no estar tan preocupados por este factor, tendrían confianza en que el medicamento estándar será adecuado para tratarlos. Creían que no tenían necesidad de probar un medicamento experimental que tal vez los expusiese a efectos secundarios no previstos. En consecuencia, los participantes con una carga viral baja (en verde) se inclinarán por inscribirse en el grupo control. Pregunta para debatir: ¿Este ensayo clínico llegará a una conclusión acertada? ¿Sí? ¿No? ¿Por qué? Respuesta: No, los grupos son desiguales y, por lo tanto, hay sesgo (parcialidad) y esto influirá en los resultados. Los grupos no son iguales, porque el grupo experimental tiene muchas más personas con cargas virales altas y esto puede afectar el resultado del estudio. = mayor carga = menor carga
 elijan participar en el grupo experimental. Al contrario, los participantes con cargas virales bajas, al no estar tan preocupados por este factor, tendrían confianza en que el medicamento estándar será adecuado para tratarlos. Creían que no tenían necesidad de probar un medicamento experimental que tal vez los expusiese a efectos secundarios no previstos. En consecuencia, los participantes con una carga viral baja (en verde) se inclinarán por inscribirse en el grupo control. Pregunta para debatir: ¿Este ensayo clínico llegará a una conclusión acertada ¿Sí ¿No ¿Por qué Respuesta: No, los grupos son desiguales y, por lo tanto, hay sesgo (parcialidad) y esto influirá en los resultados. Los grupos no son iguales, porque el grupo experimental tiene muchas más personas con cargas virales altas y esto puede afectar el resultado del estudio. = mayor carga. = menor carga.")
18
Manual del instructor para Comités de Asesoría Comunitaria
Module 2: Introduction to Clinical Trials Estudio doble ciego Enmascaramiento doble: Ni el participante ni el investigador saben la asignación de tratamiento durante todo el ensayo clínico. Se habla de enmascaramiento doble o estudios doble ciego cuando ni el participante ni el investigador sabe en qué grupo participan los sujetos. Al igual que la aleatorización, el enmascaramiento reduce el riesgo de sesgo. Por ejemplo, imaginen un estudio en el que un grupo de participantes recibe un medicamento experimental para tratar el dolor y el otro grupo un tipo de medicamento estándar. En un estudio doble ciego, ni el investigador ni el participante, sabe si el participante está asignado al grupo experimental o de control. De esta forma, no pueden estar influenciados por lo que "esperan" que suceda. Una vez que se ha obtenido toda la información en un estudio ciego, el estudio se "desenmascara" y los investigadores analizan los datos. Entonces sabrán qué medicamento es más eficaz e inocuo (seguro). Los investigadores también les comunicarán a los participantes los resultados y qué medicamento están tomando. Instructor: Para demostrar el concepto de “enmascaramiento”, asigne a todos los participantes en el grupo de capacitación sin decirles a qué grupo pertenecen. Dígales a los participantes que han sido asignados a un grupo y que cada grupo recibirá el almuerzo. Explique que los participantes del grupo 1 recibirán un almuerzo que se preparó con una receta que el cocinero nunca había usado antes, mientras que los del grupo 2 recibirán un almuerzo preparado con una receta que el cocinero ya ha usado muchas veces antes con gran éxito. Se servirá el almuerzo, pero cada participante lo llevará a su casa en vez de comerlo en grupo. Se llevarán un formulario de evaluación para calificar cuánto han disfrutado el almuerzo. Se recogerán y estudiarán los datos de los formularios de evaluación y luego se les dirá a los participantes a qué grupo se les asignó. Debate: ¿Los participantes creen que su evaluación del almuerzo podría haber sido parcial si hubieran sabido por anticipado a qué grupo se les asignaría?
. Los investigadores también les comunicarán a los participantes los resultados y qué medicamento están tomando. Instructor: Para demostrar el concepto de enmascaramiento , asigne a todos los participantes en el grupo de capacitación sin decirles a qué grupo pertenecen. Dígales a los participantes que han sido asignados a un grupo y que cada grupo recibirá el almuerzo. Explique que los participantes del grupo 1 recibirán un almuerzo que se preparó con una receta que el cocinero nunca había usado antes, mientras que los del grupo 2 recibirán un almuerzo preparado con una receta que el cocinero ya ha usado muchas veces antes con gran éxito. Se servirá el almuerzo, pero cada participante lo llevará a su casa en vez de comerlo en grupo. Se llevarán un formulario de evaluación para calificar cuánto han disfrutado el almuerzo. Se recogerán y estudiarán los datos de los formularios de evaluación y luego se les dirá a los participantes a qué grupo se les asignó. Debate: ¿Los participantes creen que su evaluación del almuerzo podría haber sido parcial si hubieran sabido por anticipado a qué grupo se les asignaría")
19
Estudios controlados con placebo
Manual del instructor para Comités de Asesoría Comunitaria Module 2: Introduction to Clinical Trials Estudios controlados con placebo ¿Qué sucede cuando no se conoce ningún medicamento eficaz como tratamiento? Los investigadores comparan el nuevo medicamento con un placebo. El placebo y el medicamento experimental se elaboran para que tengan exactamente el mismo aspecto. Grupo experimental en comparación con Grupo control con placebo Dijimos antes que para determinar si un medicamento nuevo es eficaz debemos compararlo con un medicamento que ya sepamos que es eficaz. Pero...¿qué sucede cuando no se conoce ningún medicamento eficaz como tratamiento? Por ejemplo, cuando los médicos descubrieron el SIDA por primera vez, a principios de los 80, no había tratamientos. Pasaron varios años antes de que los investigadores probaran el primer medicamento antirretroviral (ARV). Como estaban probando los primeros ARV, no tenían un medicamento para usar con el grupo control. Lamentablemente, el tratamiento estándar era que no había tratamiento para el VIH. Cuando los investigadores estudian un nuevo medicamento, y no hay otro tratamiento disponible, en general, lo comparan con un placebo, un comprimido que tiene exactamente el mismo aspecto que el medicamento nuevo pero que no contiene medicamento (es decir no tiene un principio activo). Se elabora el placebo y los comprimidos para que tengan exactamente el mismo aspecto. De esa forma, los investigadores y los participantes no saben quién toma el comprimido con medicamento o quién toma el placebo. Y así se reduce el riesgo de sesgo. Cuando el grupo control recibe placebo, el estudio se llama estudio controlado con placebo.
. Como estaban probando los primeros ARV, no tenían un medicamento para usar con el grupo control. Lamentablemente, el tratamiento estándar era que no había tratamiento para el VIH. Cuando los investigadores estudian un nuevo medicamento, y no hay otro tratamiento disponible, en general, lo comparan con un placebo, un comprimido que tiene exactamente el mismo aspecto que el medicamento nuevo pero que no contiene medicamento (es decir no tiene un principio activo). Se elabora el placebo y los comprimidos para que tengan exactamente el mismo aspecto. De esa forma, los investigadores y los participantes no saben quién toma el comprimido con medicamento o quién toma el placebo. Y así se reduce el riesgo de sesgo. Cuando el grupo control recibe placebo, el estudio se llama estudio controlado con placebo.")
20
Estudio controlado con placebo
Manual del instructor para Comités de Asesoría Comunitaria Module 2: Introduction to Clinical Trials Estudio controlado con placebo Instructor: Si desea demostrar el concepto de placebo, muestre 2 botellas de agua que tengan exactamente el mismo aspecto. Diga al grupo que ha agregado una pequeña cantidad de sal a una de las botellas, y que esta agua salada representa el medicamento experimental, mientras que el agua sin sal representa al grupo controlado con placebo. Pídales que imaginen que se les entrega una de las botellas (sin saber qué tipo de botella es) y una hora después de beber el agua, se les pide que informen cuánta sed tienen. ¿Les parece que su evaluación de la sed estaría influenciada si hubieran sabido qué tipo de agua recibían? Ahora, pida a los participantes que imaginen que la mitad de la clase recibe una botella de agua (el grupo experimental) y que la otra mitad no recibe nada. Pregunte, "¿Saber que han recibido o no han recibido agua influiría en su percepción de cuánta sed creen que tienen?" experimental control
 y una hora después de beber el agua, se les pide que informen cuánta sed tienen. ¿Les parece que su evaluación de la sed estaría influenciada si hubieran sabido qué tipo de agua recibían Ahora, pida a los participantes que imaginen que la mitad de la clase recibe una botella de agua (el grupo experimental) y que la otra mitad no recibe nada. Pregunte, ¿Saber que han recibido o no han recibido agua influiría en su percepción de cuánta sed creen que tienen experimental. control.")
21
Ética de los estudios controlados con placebo
Manual del instructor para Comités de Asesoría Comunitaria Module 2: Introduction to Clinical Trials Ética de los estudios controlados con placebo Si se dispone de un tratamiento, el estudio no puede por motivos éticos, asignar a un grupo a recibir placebo sin ningún tratamiento. Pero si no se conoce tratamiento, el estudio puede éticamente incluir un grupo control que recibirá placebo. La razón es que el grupo placebo recibirá el mismo tratamiento que recibiría si no participara en el estudio (tratamiento estándar). Hay estrictas normas internacionales sobre las investigaciones clínicas para asegurarse de que sean éticas (justas, equitativas y correctas). En el módulo 6 de la capacitación hablaremos en profundidad de la ética del uso de placebo en los ensayos clínicos. En pocas palabras, si hay un tratamiento disponible no es ético que haya un grupo que reciba sólo tratamiento, es decir, solamente placebo. Si no hay tratamiento disponibles, no hay tratamiento estándar y entonces sí es ético usar un grupo placebo para compararlo con el tratamiento experimental.
. Hay estrictas normas internacionales sobre las investigaciones clínicas para asegurarse de que sean éticas (justas, equitativas y correctas). En el módulo 6 de la capacitación hablaremos en profundidad de la ética del uso de placebo en los ensayos clínicos. En pocas palabras, si hay un tratamiento disponible no es ético que haya un grupo que reciba sólo tratamiento, es decir, solamente placebo. Si no hay tratamiento disponibles, no hay tratamiento estándar y entonces sí es ético usar un grupo placebo para compararlo con el tratamiento experimental.")
22
Regulación de cuestiones éticas
Declaración de Helsinki Normas de “Buena Práctica Clínica” en Internacional Conference for Harmonisation (ICH). Los países han adaptado su legislación a estas normas
. Los países han adaptado su legislación a estas normas.")
23
Legislación española La ley 25/1990 del Medicamento Real Decreto 561/1993 Normas de Buena Práctica Clínica (BPC). "Procedimientos Normalizados de Trabajo"
. Procedimientos Normalizados de Trabajo")
24
Ley 25/1990 del Medicamento EC: evaluación experimental de una sustancia a través de su aplicación a seres humanos. Efectos farmacodinámicos Establecer eficacia (terapéutica, profiláctica o diagnóstica). Conocer reacciones adversas y establecer su seguridad
. Conocer reacciones adversas y establecer su seguridad.")
25
Normas de Buena Práctica Clínica (BPC).
Procedimientos (éticos y científicos) exigidos en EC Aseguran la existencia de medicamentos seguros, de calidad y eficaces.
 exigidos en EC. Aseguran la existencia de medicamentos. seguros, de calidad. y eficaces.")
26
Pilares Buena Práctica Clínica
Protección del sujeto según los mas altos estándares científicos y éticos Adopción de los Procedimientos Normalizados de Trabajo (PNT) Archivo completo Adecuada notificación de las reacciones adversas (GCP for trials on Medicinal Product in the European Community. July 1990).
 Archivo completo. Adecuada notificación de las reacciones adversas. (GCP for trials on Medicinal Product in the European Community. July 1990).")
27
Real Decreto 561/1993 Requisitos para la realización de E. C.
El título III regula a los Comités Éticos de Investigación Clínica (CEIC): 1. Mínimo 7 miembros (2 ajenos sanidad ~ Lic Derecho). 2. Composición: médicos (1 Farm. Clinico), Farmacéutico y enfermera. 4. No podrán percibir remuneración del promotor del ensayo.
: 1. Mínimo 7 miembros (2 ajenos sanidad ~ Lic Derecho). 2. Composición: médicos (1 Farm. Clinico), Farmacéutico y enfermera. 4. No podrán percibir remuneración del promotor del ensayo.")
28
Funciones CEIC Riesgos Beneficios. Protocolo Metodológicos Éticos
Legales Riesgos Beneficios.

29
Funciones CEIC: Evaluación EC
Justificación riesgos/beneficios Idoneidad del equipo investigador Obligaciones asistenciales y otras investigaciones

30
Funciones CEIC: Evaluación EC
Información escrita sobre EC Información y consentimiento Compensación en caso de lesión atribuible al EC

31
Funciones CEIC: Evaluación EC
Conocerá y evaluará la compensación económica del promotor. Seguimiento del EC desde su inicio hasta la recepción del informe final.

32
Normas de funcionamiento
Principio de confidencialidad Mitad más uno de sus miembros Comisión de Investigación o Comité de Ética Asistencial : presencia Asesoramiento de expertos CEIC. Comunicación SAEs

33
) Problemas y dificultades Selección miembros Falta de formación
Exceso de trabajo y burocracia Gran número de protocolos Ausencia de reconocimiento Responsabilidad añadida a su labor asistencial …… )
")
34
Objetivos de hoy ¿Qué es un comité de ética en investigación clínica? Un ensayo clínico: Ensayos clínicos en oncología

35
Ensayos clínicos en oncología

36
Fase II - IV en hospitales o centro de salud

37
Al lado del investigador
Gestión de todos los aspectos: técnicos, logísticos organizativos.

38
Coordinando EECC Oncológicos
Funciones propias de enfermería. Funciones organizativo - administrativas. Funciones logísticas. Funciones técnicas.

39
Funciones propias Relaciones distintas > distintas funciones. .

40
Funciones propias Cuidados

41
Funciones organizativo-administrativas
Gestión del Archivo del Investigador Cuaderno de Recogida de Datos Gestión entradas – salidas medicación Gestión material de estudio Cambios relevantes en el protocolo.

42
Funciones logísticas Gestión recepción - devolución medicación
Gestión envío de muestras biológicas: PK/Inmunogenia Biomarcadores Tejidos Gestión otros materiales de estudio: RX…datos…

43
Funciones técnicas Obtención de muestras biológicas
Obtención de constantes vitales Administración de tests y cuestionarios Aplicación de procedimientos del estudio, Procedimientos especiales (TAC, ECG, espirometrías, pruebas de esfuerzo, etc.)
")
44
Eventos adversos graves
Muerte, evento que amenaza la vida, reingreso. Comunicación 48 h Formularios de informes de reacciones adversas graves Carpeta de reglamentación con documentación relacionada con el estudio..

45
Inclusión de Pacientes
Criterios Inclusión/Exclusión Estudio y revisión minuciosa Firma consentimiento

46
Información al paciente
Revisión del protocolo Cronograma terapéutico Cronograma pruebas Cronograma Control de PE Cronograma Revisiones

47
Controles del Estudio Revisiones CRD periódicas de todos los registros por el patrocinador Cortes de datos periódicos multicéntricos AEM y FDA

55
En la actualidad, un ensayo clínico farmacológico es toda evaluación experimental de una sustancia o fármaco, a través de su administración o aplicación a seres humanos, orientada a alguno de los siguientes fines: Poner de manifiesto sus efectos farmacodinámicos o recoger datos referentes a su absorción, distribución, metabolismo y excreción en el organismo humano. Establecer la eficacia para una indicación terapéutica, profiláctica o diagnóstica determinada. Conocer el perfil de sus reacciones adversas y establecer su seguridad.

56
Un investigador supervisa el estudio en su conjunto
Un investigador supervisa el estudio en su conjunto. Como coordinador de la investigación, que mantenga el proyecto actualmente en marcha a lo largo de la coordinación de los esfuerzos de los involucrados, incluidos los investigadores principales, otros miembros del equipo (por ejemplo, enfermería, farmacia, laboratorio, radiología y personal), los pacientes y sus familias, la tramitación de documentos normativos y financieros , y trabajando con el investigador en la supervisión de la selección y el reclutamiento de pacientes y la ejecución del protocolo del estudio.
, los pacientes y sus familias, la tramitación de documentos normativos y financieros , y trabajando con el investigador en la supervisión de la selección y el reclutamiento de pacientes y la ejecución del protocolo del estudio.")
57
Una vez que el IRB concede la aprobación (que generalmente toma un par de semanas), usted y el investigador puede comenzar la investigación, la contratación, y la inclusión de pacientes, un largo proceso que asegura una base sólida para el estudio y sus resultados. historias clínicas y de laboratorio y radiología resultados ayudarán a determinar qué pacientes son candidatos apropiados.

58
El médico que supervisa el estudio o un miembro del equipo de estudio debe obtener el consentimiento informado de cada paciente (o su sustituto) después de su examen, pero antes de inscribirse al paciente e iniciar los procedimientos de estudio. Para cada paciente inscrito, usted mantener una forma de recogida de datos detallados (CRF), que incluye la historia clínica del paciente, la información inicial de otros, y las actualizaciones diarias para seguir el progreso del paciente, los reveses y los resultados.
, que incluye la historia clínica del paciente, la información inicial de otros, y las actualizaciones diarias para seguir el progreso del paciente, los reveses y los resultados..")
59
Para los datos del estudio preciso, el protocolo debe ser seguido escrupulosamente. Debido a que el personal de enfermería juega un papel tan central en la ejecución del protocolo, que necesita para garantizar (a través de los programas de desarrollo personal) que saben cómo llevar a cabo todas las tareas requeridas por el protocolo. Una vez que el protocolo se inicia, se le supervisará la ejecución de cerca. Usted puede incluso ayudar a llevar a cabo el protocolo mismo, para evitar sobrecargar al personal de enfermería.
 que saben cómo llevar a cabo todas las tareas requeridas por el protocolo. Una vez que el protocolo se inicia, se le supervisará la ejecución de cerca. Usted puede incluso ayudar a llevar a cabo el protocolo mismo, para evitar sobrecargar al personal de enfermería..")
60
Muestras sangre

61
Controles rx

62
Los eventos adversos graves (AAG), como la muerte, un evento que amenaza la vida, o reingreso, deben ser reportados a la IRB. Si un estudio patrocinado por la industria está llevando a cabo en varios centros, usted tiene que reportar reacciones adversas graves que ocurren en cualquiera de los sitios. Los formularios de informes de reacciones adversas graves requieren una amplia documentación del evento. También debe informar a la ninguna modificación en el estudio o su personal IRB. Usted mantener una carpeta de reglamentación que incluye la documentación relacionada con el estudio. En un estudio multicéntrico, puede ser necesario personal adicional para mantenerse en la cima de trámites reglamentarios.

63
Inscripción de Pacientes:
La revisión es el primer paso para la inclusión de pacientes en un estudio y consiste en revisar historias clínicas, resultados de laboratorio, radiología y los resultados para determinar si el paciente cumple los criterios para la inscripción. El proceso de selección es a menudo mucho tiempo, pero es un componente importante para asegurar la inscripción de pacientes e integridad de los resultados del estudio. Después de la inscripción comienza, el coordinador de la investigación debe mantener el IRB informó de efectos adversos graves (SAE) que se producen con el estudio y los pacientes del estudio. Si un estudio es patrocinado por la industria con la inscripción multicéntrico, la IRB también quiere saber de que ocurran fuera del SAE de la institución, así como las recomendaciones formuladas por la Junta de Control de Seguridad de Datos (DSMB) asignado para revisar el estudio. El proceso de SAE consiste en un formato normalizado (s) generado por la oficina del IRB y de la industria de los patrocinadores del estudio (cuando corresponda). Se recopila información sobre la probabilidad de que el SAE está causada por un procedimiento relacionado con el estudio, la medicación o tratamiento. Si el estudio es patrocinado por la industria e implica un medicamento o dispositivo, el informe de la SAE se requiere dentro de las 24 horas del equipo de estudio de la notificación del evento. Si el estudio no es patrocinado por la industria, la oficina del IRB por lo general se establecen directrices para los requisitos de notificación. En cada caso, la documentación de apoyo, incluidos los
 que se producen con el estudio y los pacientes del estudio. Si un estudio es patrocinado por la industria con la inscripción multicéntrico, la IRB también quiere saber de que ocurran fuera del SAE de la institución, así como las recomendaciones formuladas por la Junta de Control de Seguridad de Datos (DSMB) asignado para revisar el estudio. El proceso de SAE consiste en un formato normalizado (s) generado por la oficina del IRB y de la industria de los patrocinadores del estudio (cuando corresponda). Se recopila información sobre la probabilidad de que el SAE está causada por un procedimiento relacionado con el estudio, la medicación o tratamiento. Si el estudio es patrocinado por la industria e implica un medicamento o dispositivo, el informe de la SAE se requiere dentro de las 24 horas del equipo de estudio de la notificación del evento. Si el estudio no es patrocinado por la industria, la oficina del IRB por lo general se establecen directrices para los requisitos de notificación. En cada caso, la documentación de apoyo, incluidos los.")
64
El coordinador del estudio es también la obligación de informar las modificaciones a la IRB de manera oportuna, incluyendo la adición o eliminación de personal del estudio, las modificaciones del protocolo, los cambios en el consentimiento, el aumento de las solicitudes de inscripción, la adición de nuevos procedimientos, añadir o eliminar un sitio, los cambios en la financiación, y aprobaciones de publicidad. Si su sitio está participando en múltiples estudios, tanto de los NIH y patrocinado por la industria, el personal adicional puede ser necesaria para mantenerse al día con toda la documentación de reglamentación. Consulte la tabla 2 para una lista de importantes agencias reguladoras y sus enlaces a las webs.

65
Un asociado de investigación clínica-patrocinador del empleado vendrá a revisar la FRC-incluido el control contra registros médicos del paciente y ver que tu carpeta de reglamentación se ha completado. Usted necesitará para corregir cualquier discrepancia en el FRC. Este largo proceso es esencial que el estudio nunca someterse a una auditoría por parte de la Administración de Alimentos y Medicamentos. Cuando el estudio es más, mantenga todos los documentos relacionados con el estudio de forma segura su confidencialidad. Aunque usted debe tener una lista de nombres de los pacientes asegurados y la información de contacto, las regulaciones de HIPAA requieren que la información de identificación se eliminan de los datos del estudio antes de tomar los datos disponibles para el análisis.

68
En conclusión, el papel de un coordinador de la investigación de cuidados intensivos pueden ser muy complejos, que requieren el coordinador de aprovechar la experiencia previa y conocimientos especializados en la UCI. Además de su experiencia clínica y la experiencia, el coordinador también debe poseer una personalidad organizada y detallista para navegar por la evolución de las necesidades de reglamentación. Encontrar la persona adecuada para esta posición ayudará a asegurar que la investigación clínica de calidad se está realizando en su institución, así como proporcionar una carrera productiva y gratificante para el coordinador de la investigación.

70
HASTA AKIIIIIIIIIIIIIIIIIIIIII

71
Ensayos clínicos Evaluación experimental de un producto, sustancia medicamento, técnica diagnóstica o terapéutica que, en su aplicación a seres humanos, pretende valorar su eficacia y seguridad.

72
Los estudios de prometedores tratamientos nuevos o experimentales en pacientes se conocen como ensayos clínicos. Un ensayo clínico se realiza sólo cuando hay razones para creer que el tratamiento que se está estudiando puede ser beneficioso para el paciente. Los tratamientos usados en los ensayos clínicos con frecuencia demuestran tener beneficios reales. Los investigadores realizan estudios sobre nuevos tratamientos para conocer la utilidad del nuevo tratamiento, el mecanismo de acción del nuevo tratamiento, si la efectividad es mayor que otros tratamientos ya disponibles, los efectos secundarios del nuevo tratamiento y si son mayores o menores que el tratamiento convencional, si supera los beneficios a los efectos secundarios y en qué pacientes el nuevo tratamiento es más útil.

73
Origen de los nuevos fármacos
Existen cinco fuentes principales de nuevos fármacos: [editar] Diseño racional de moléculas Se trata del diseño sobre la base de la comprensión del mecanismo patogénico de la enfermedad a nivel molecular. Uno de los casos más notables es el del diseño de las oximas para el tratamiento de la intoxicación por inhibidores de la colinesterasa, ya que estos agentes se derivaron del conocimiento del sitio de la acción tóxica, así como del tipo de interacción química que se presentaba en ese caso; en cierta forma, cuando se descubrió que los antipsicóticos más eficaces bloqueaban a los receptores D2 y 5HT2A, pudieron diseñarse agentes que bloquearan tales receptores. Otro ejemplo es el de la obtención de las estatinas a partir del conocimiento de la molécula del Acetil-coenzima-A. Aunque se trata de la fuente “ideal” de nuevos fármacos, es también una de las menos eficientes, ya que aún el conocimiento sobre las bases de la enfermedad humana son incompletos. [editar] Estudio de Moléculas creadas al azar Aunque pudiera parecer lo contrario, este es uno de los métodos más provechosos para la obtención de fármacos nuevos, sobre todo desde el punto de vista económico. Con relación a los grandes costos de la obtención de un nuevo fármaco, el proceso de síntesis química “al azar” (sumado al de la realización de pruebas preliminares de la acción de los compuestos resultantes) resulta sumamente barato. [editar] Tamizaje de productos naturales Aunque los primeros agentes farmacológicos realmente eficaces fueron obtenidos de fuentes naturales (digitálicos, penicilina, opioides, insulina, etc.), el tamizaje de productos naturales fue hasta cierto punto dejado de lado por un tiempo considerable. Sin embargo, esta metodología ha vuelto a ganar adeptos y se han obtenido moléculas que representan verdaderas novedades en campos diversos, como la terapia de las enfermedades neoplásicas (taxol) o el tratamiento de la malaria (artemisininas). Aunque cada vez aumenta el número de fármacos de otros orígenes, todavía en la actualidad alrededor de un 40% de los nuevos agentes deriva de productos naturales (más de la mitad de los cuales son plantas). [editar] Modificación de moléculas conocidas Este procedimiento en teoría debería limitarse al mejoramiento de la eficacia y o seguridad de fármacos ya existentes, pero en realidad se utiliza básicamente para la obtención de nuevas moléculas que puedan soslayar los derechos de patente de otras que ya hayan ganado cierto espacio en el mercado farmacéutico (me too drugs). [editar] Biotecnología Consiste fundamentalmente en el uso de métodos de clonación genética para la obtención de ciertas moléculas peptídicas. Solo una de cada a nuevas moléculas evaluadas inicialmente llega a pasar por todo el proceso de evaluación de nuevos fármacos, aunque esto no garantiza que la misma sea finalmente comercializada, y, si es comercializada, no garantiza su persistencia ulterior en el mercado.
 resulta sumamente barato. [editar] Tamizaje de productos naturales. Aunque los primeros agentes farmacológicos realmente eficaces fueron obtenidos de fuentes naturales (digitálicos, penicilina, opioides, insulina, etc.), el tamizaje de productos naturales fue hasta cierto punto dejado de lado por un tiempo considerable. Sin embargo, esta metodología ha vuelto a ganar adeptos y se han obtenido moléculas que representan verdaderas novedades en campos diversos, como la terapia de las enfermedades neoplásicas (taxol) o el tratamiento de la malaria (artemisininas). Aunque cada vez aumenta el número de fármacos de otros orígenes, todavía en la actualidad alrededor de un 40% de los nuevos agentes deriva de productos naturales (más de la mitad de los cuales son plantas). [editar] Modificación de moléculas conocidas. Este procedimiento en teoría debería limitarse al mejoramiento de la eficacia y o seguridad de fármacos ya existentes, pero en realidad se utiliza básicamente para la obtención de nuevas moléculas que puedan soslayar los derechos de patente de otras que ya hayan ganado cierto espacio en el mercado farmacéutico (me too drugs). [editar] Biotecnología. Consiste fundamentalmente en el uso de métodos de clonación genética para la obtención de ciertas moléculas peptídicas. Solo una de cada a nuevas moléculas evaluadas inicialmente llega a pasar por todo el proceso de evaluación de nuevos fármacos, aunque esto no garantiza que la misma sea finalmente comercializada, y, si es comercializada, no garantiza su persistencia ulterior en el mercado.")
74
Ensayo clínico farmacológico
Dado que los animales de laboratorio se presentan como cepas con variabilidad biológica limitada, los estudios realizados con ellos no pueden ser suficientes para determinar sin dudas que un fármaco determinado tendrá las características deseadas de eficacia y seguridad en poblaciones humanas; esto no solo depende de las diferencias entre las especies, sino también de la posibilidad de reacciones que no pueden ser adecuadamente determinadas en animales (cefalea, depresión, tinitus, etc.). Por esta razón, antes de su posible aprobación, un fármaco debe ser probado en seres humanos, a través de una metodología que distingue tres fases, considerando el estudio y seguimiento de un fármaco después de su comercialización como una cuarta fase. [editar] Estudios clínicos fase I Representa la primera administración en humanos, generalmente en pequeño número, que rara vez es mayor de 100. Para esta fase, la administración se realiza generalmente en adultos jóvenes sanos de sexo masculino, con el fin de detectar posibles signos incipientes de toxicidad, lo que permitiría determinar luego el rango seguro de dosificación. Los aspectos farmacocinéticos se suelen medir también, aunque su estudio no es el objetivo principal de esta fase. [editar] Estudios clínicos fase II Si la comprobación preliminar de seguridad en la fase I ha sido satisfactoria, se pasa a esta fase, la cual involucra la administración del fármaco a individuos que presentan la enfermedad para la que se ha concebido su empleo. Este grupo de pacientes debe ser relativamente homogéneo en sus características basales (presentar solo la enfermedad en cuestión) y no se suelen incluir más de 100 a 200 individuos (a veces, con los mejores medicamentos disponibles para el tratamiento de la enfermedad implicada y si tales fármacos no existen, la comparación sería con placebo). La finalidad de la fase II es la de establecer mediciones preliminares de la relación eficacia terapéutica/toxicidad, así como establecer la dosis óptima o sus límites de variación en la condición a tratar. [editar] Estudios clínicos fase III Si se obtiene razonable evidencia de las fases I y II, comienzan los estudios de fase III, que pueden involucrar múltiples médicos tratando cientos o incluso miles de pacientes. Aparte de verificar la eficacia del medicamento, se busca determinar manifestaciones de toxicidad previamente no detectadas. En esta fase se obtiene una mejor perspectiva de la relación entre seguridad y eficacia, parámetros que han de cuantificarse en el contexto del desorden que se pretenda tratar. [editar] Estudios clínicos fase IV También conocidos como estudios de farmacovigilancia consisten en el seguimiento del fármaco después de que ha sido comercializado. Se busca básicamente la detección de toxicidad previamente insospechada, así como de la evaluación de la eficacia a largo plazo. En la fase IV se pueden detectar reacciones adversas raras, mientras que en las fases previas es excepcional el descubrimiento de aquellas con frecuencia menor a 1/1000.
. Por esta razón, antes de su posible aprobación, un fármaco debe ser probado en seres humanos, a través de una metodología que distingue tres fases, considerando el estudio y seguimiento de un fármaco después de su comercialización como una cuarta fase. [editar] Estudios clínicos fase I. Representa la primera administración en humanos, generalmente en pequeño número, que rara vez es mayor de 100. Para esta fase, la administración se realiza generalmente en adultos jóvenes sanos de sexo masculino, con el fin de detectar posibles signos incipientes de toxicidad, lo que permitiría determinar luego el rango seguro de dosificación. Los aspectos farmacocinéticos se suelen medir también, aunque su estudio no es el objetivo principal de esta fase. [editar] Estudios clínicos fase II. Si la comprobación preliminar de seguridad en la fase I ha sido satisfactoria, se pasa a esta fase, la cual involucra la administración del fármaco a individuos que presentan la enfermedad para la que se ha concebido su empleo. Este grupo de pacientes debe ser relativamente homogéneo en sus características basales (presentar solo la enfermedad en cuestión) y no se suelen incluir más de 100 a 200 individuos (a veces, con los mejores medicamentos disponibles para el tratamiento de la enfermedad implicada y si tales fármacos no existen, la comparación sería con placebo). La finalidad de la fase II es la de establecer mediciones preliminares de la relación eficacia terapéutica/toxicidad, así como establecer la dosis óptima o sus límites de variación en la condición a tratar. [editar] Estudios clínicos fase III. Si se obtiene razonable evidencia de las fases I y II, comienzan los estudios de fase III, que pueden involucrar múltiples médicos tratando cientos o incluso miles de pacientes. Aparte de verificar la eficacia del medicamento, se busca determinar manifestaciones de toxicidad previamente no detectadas. En esta fase se obtiene una mejor perspectiva de la relación entre seguridad y eficacia, parámetros que han de cuantificarse en el contexto del desorden que se pretenda tratar. [editar] Estudios clínicos fase IV. También conocidos como estudios de farmacovigilancia consisten en el seguimiento del fármaco después de que ha sido comercializado. Se busca básicamente la detección de toxicidad previamente insospechada, así como de la evaluación de la eficacia a largo plazo. En la fase IV se pueden detectar reacciones adversas raras, mientras que en las fases previas es excepcional el descubrimiento de aquellas con frecuencia menor a 1/1000.")
75
Fases de lnvestigación Clínica Fases I a IV
Dr. Armando González Jaimes AstraZeneca

77
Investigación Clínica Farmacéutica Desarrollo de Estudios Clínicos
El desarrollo de los estudios clínicos para demostrar la eficacia y la seguridad de una nueva droga, responden a los requerimientos del plan de desarrollo farmacéutico de un nuevo compuesto o grupo de compuestos de una Compañía

78
Investigación Clínica Farmacéutica Desarrollo de Estudios Clínicos
¿Como se determina el plan de desarrollo farmacéutico? Status mundial de una enfermedad común Tendencia de la enfermedad en el futuro Estado actual del mercado farmacéutico para esa enfermedad Factibilidad para realizarse dentro de la Compañía Decisión ejecutiva

79
Investigación Clínica Farmacéutica Desarrollo de Estudios Clínicos
Pasos a seguir: Buscar información específica sobre la enfermedad Estructurar una meta con enfoque mercadológico Estudiar la estructura química de los compuestos partiendo de una base de datos farmacológica Se analizan los efectos, el metabolismo y la toxicología de un grupo de compuestos Se traslapan los modelos de la enfermedad con las moléculas desarrolladas para la implementación de estudios clínicos

80
Investigación Clínica Farmacéutica Desarrollo de Estudios Clínicos
Los objetivos claves son crear una medicamento que reúna las siguientes propiedades: Seguro y Eficaz La autoridad regulatoria lo apruebe El paciente lo pida El doctor lo prescriba

81
Investigación Clínica Farmacéutica Desarrollo de Estudios Clínicos
El plan de desarrollo se divide en FASES PRECLINICAS FASES CLINICAS

82
Investigación Clínica Farmacéutica Desarrollo de Estudios Clínicos
El proceso de desarrollo de medicamentos comprende inicialmente fases preclínicas Fase de búsqueda documental Fase de pruebas en modelos cibernéticos Fase de síntesis en laboratorios de química Fase de farmacología preclínica en modelos animales Fase de toxicología preclínica

83
Investigación Clínica Farmacéutica Desarrollo de Estudios Clínicos
Fases Clínicas El propósito principal de cualquier estudio clínico realizado por una compañía farmacéutica es evaluar un régimen terapéutico, al mismo tiempo que tratar una enfermedad

84
Investigación Clínica Farmacéutica Desarrollo de Estudios Clínicos
Las razones comunes para diseñar y realizar un estudio clínico son: Establecer la seguridad de un nuevo fármaco Determinar la eficacia y la dosis óptima Determinar la farmacología Demostrar la seguridad y eficacia en una población más amplia Comparar la eficacia de un nuevo compuesto contra los tratamientos convencionales Obtener o evitar la inclusión de textos específicos en la monografía del medicamento Estudiar grupos específicos de pacientes Los requerimientos regulatorios

85
Investigación Clínica Farmacéutica Desarrollo de Estudios Clínicos
La elección del período de tratamiento depende de: La duración del período de la enfermedad (aguda, crónica, intermitente) La farmacocinética y la farmacodinamia El grado de cooperación requerido por parte de los pacientes
 La farmacocinética y la farmacodinamia. El grado de cooperación requerido por parte de los pacientes.")
86
Investigación Clínica Farmacéutica Desarrollo de Estudios Clínicos
Para cualquier estudio de investigación clínica, la fase del estudio en el proceso de desarrollo clínico definirá los aspectos del diseño del estudio, incluyendo la población de pacientes, el tamaño de la muestra y la duración del tratamiento

87
Investigación Clínica Farmacéutica Desarrollo de Estudios Clínicos
Fases Clínicas Fase I Fase II Fase III Fase IV TODAS LAS FASES CLINICAS BUSCAN DETERMINAR SEGURIDAD Y EFICACIA

89
Investigación Clínica Farmacéutica
FASE I Su objetivo es evaluar su efecto principal en el órgano blanco, determinar la dosis efectiva, y corrobar sus propiedades farmacológicas. Determinación de valores farmacocinéticos y farmacodinámicos Los participantes son voluntarios sanos en la mayoría de los casos. El diseño del estudio es simple. Generalmente se desarrollan en un solo centro de investigación que cuente con una unidad especializada para monitoreo no invasivo/invasivo y laboratorios de análisis farmacológico. De seis meses a dos años 1 - 8 moléculas probadas

91
Investigación Clínica Farmacéutica
FASE II Corroborar el efecto deseado en el órgano blanco (enfermedad) Corroborar/evaluar los efectos en otros órganos/ sistemas. Los participantes son pacientes. Se compara la nueva droga con una similar ya comercializada o contra placebo Se ajusta/confirma la dosis seleccionada Pueden aplicarse cuestionarios de calidad de vida, farmacoeconomía El diseño es complejo Generalmente son multicéntricos De 2 a 3 años Hasta 3 moléculas
 Corroborar/evaluar los efectos en otros órganos/ sistemas. Los participantes son pacientes. Se compara la nueva droga con una similar ya comercializada o contra placebo. Se ajusta/confirma la dosis seleccionada. Pueden aplicarse cuestionarios de calidad de vida, farmacoeconomía. El diseño es complejo. Generalmente son multicéntricos. De 2 a 3 años. Hasta 3 moléculas.")
93
Investigación Clínica Farmacéutica
FASE III Más dirigidos a eficacia que a seguridad Comprobar la eficacia en la(s) dosis elegidas Probar una o varias hipótesis primarias Son los estudios que se utilizan para solicitar registro a las autoridades sanitarias Se obtiene información de calidad de vida y economía de la salud Multicéntricos De 3 a 4 años 1 o 2 moléculas
 dosis elegidas. Probar una o varias hipótesis primarias. Son los estudios que se utilizan para solicitar registro a las autoridades sanitarias. Se obtiene información de calidad de vida y economía de la salud. Multicéntricos. De 3 a 4 años. 1 o 2 moléculas.")
95
Investigación Clínica Farmacéutica
FASE IV Se realizan después que el producto ha conseguido permiso para comercialización Enriquecen el perfil de seguridad del producto Familiarizan a los médicos con el fármaco Cada vez son más frecuentemente requeridos por las autoridades regulatorias Son usados como extensión de los estudios de fase III para recabar datos de seguridad con el uso prolongado Ampliamente multicéntricos Hasta 6 años

98
Ensayos clínicos en oncología

99
COORDINADOR de ENSAYOS CLINICOS para colaborar en sus ensayos en la zona de Elche. Su misión consistirá en asistir al equipo investigador para supervisar el progreso de un ensayo clínico para que el mismo sea realizado, registrado e informado de acuerdo con el protocolo, los Procedimientos Normalizados de Trabajo (PNTs), la Buena Práctica Clínica (BPC) y los requisitos reguladores pertinentes.
, la Buena Práctica Clínica (BPC) y los requisitos reguladores pertinentes..")
Presentaciones similares



















 ¿Quién es Betty C Jung? Revise.>")










![M K Unnikrishnan [Agosto 2006]](/3/1105489/big_thumb.jpg "M K Unnikrishnan [Agosto 2006]>")




