Descargar la presentación
La descarga está en progreso. Por favor, espere

1
Manejo de Intoxicaciones en Pediatría

2
Manejo de Intoxicaciones en Pediatría

3
Dr. Angelo Lopez_ Pediatra intensivista HUSI. Epidemiólogo

4
Intoxicación Cuadro clínico producido por el contacto con una sustancia tóxica o veneno, que ingresa al organismo produciendo alteraciones patológicas en el mismo. Peña, Guia para el manejo del paciente intoxicado.Dpto de farmacología y toxicología. Universidad de Antioquia

5
Tóxico Sustancia que es susceptible de generar, por un mecanismo químico, acciones adversas o nocivas en los seres vivos. Depende de: Dosis Características del organismo receptor (raza, sexo, edad, condiciones fisiológicas y patológicas). Características externas: ambiente, temperatura, situación atmosférica. Vía de administración Peña, Guia para el manejo del paciente intoxicado.Dpto de farmacología y toxicología. Universidad de Antioquia
. Características externas: ambiente, temperatura, situación atmosférica. Vía de administración. Peña, Guia para el manejo del paciente intoxicado.Dpto de farmacología y toxicología. Universidad de Antioquia")
6
Término de uso legal o jurídico, no médico.
Veneno Término de uso legal o jurídico, no médico. El concepto implica las sustancias tóxicas que son empleadas de manera intencional. Envenenamiento sólo cuando las intoxicaciones son homicidas o suicidas. Peña, Guia para el manejo del paciente intoxicado.Dpto de farmacología y toxicología. Universidad de Antioquia

7
Dosis DOSIS: Es la cantidad de tóxico que ingresa al organismo y que es capaz de producir síntomas de toxicidad DOSIS TOXICA: dosis que es capaz de producir los efectos dañinos DOSIS UMBRAL: cantidad mínima necesaria para producir el efecto tóxico. Peña, Guia para el manejo del paciente intoxicado.Dpto de farmacología y toxicología. Universidad de Antioquia

8
Toxidrome Signos y síntomas presentados por el paciente con un grupo de agentes tóxicos de mecanismos de toxicidad similar, con el objetivo de orientar el diagnóstico, y el tratamiento antidotal y de soporte. Peña, Guia para el manejo del paciente intoxicado.Dpto de farmacología y toxicología. Universidad de Antioquia

9
Generalidades 99% Intoxicaciones agudas 92% ocurren en el domicilio
40% por medicamentos 95% auto-ingestión 92% ocurren en el domicilio 88% accidentales 62% niños menores de 6 años Watson WA, Litovitz TL, Rodgers GCJ, et al Annual report of the American Association of Poison Control Centers Toxic Exposure Surveillance System. Am J Emerg Med 2005;23:589–666. David L. Eldridge. Pediatric Toxicology. Emerg Med Clin N Am 15 (2007) 283–308
 283–308.")
10
Epidemiología USA: 2004 American Association of Poison Control Centers (AAPCC) 1,250,536 exposiciones a sustancias tóxicas (51, 3 % ) menores de 6 años y 938,874 (38,5%) menores de 2 años. Muerte rara. 15,447 de las cuales 537 (3,7%) en menores de 6 años y 397 (2,6%) en menores de 2 años. David L. Eldridge. Pediatric Toxicology. Emerg Med Clin N Am 15 (2007) 283–308
 1,250,536 exposiciones a sustancias tóxicas (51, 3 % ) menores de 6 años y 938,874 (38,5%) menores de 2 años. Muerte rara. 15,447 de las cuales 537 (3,7%) en menores de 6 años y 397 (2,6%) en menores de 2 años. David L. Eldridge. Pediatric Toxicology. Emerg Med Clin N Am 15 (2007) 283–308.")
11
Hanhan, Usama. The poisoned child in the pediatrica Intensive Care Unit. Pediatr Clin N Am 55 (2008)
")
12
Epidemiología COLOMBIA : SIVIGILA INS Intoxicaciones . Exposición a plaguicidas 45,2%, medicamentos 21,1%. exposiciones . Tasa de intoxicaciones 5,75 x habitantes. 28,4% menores de 14 años; 9,3% menores de 4 años. Muerte rara casos. Tasa de mortalidad 0,6 x habitantes// Tasa de letalidad 9,48 x habitantes Avila Albert; Moreno Atilio. Medidas generales en el servicio de urgencias para el paciente pediátrico intoxicado. Trabajo de promoción. Medicina de Urgencias. Pontificia Universidad Javeriana ( pendiente publicación)
")
13
Puntos Clave

14
Goldfranks, Toxicologic Emergencies, Mc Graw Hill, 7 Ed.
Anamnesis 1 Medicación sospechosa Hora del contacto Vía de intoxicación Otras medicaciones probables asociadas Antecedentes personales Bryant, Sean. Management of toxic exposure in children. Emerg Med Clin N Am 21 (2003) Goldfranks, Toxicologic Emergencies, Mc Graw Hill, 7 Ed.
 Goldfranks, Toxicologic Emergencies, Mc Graw Hill, 7 Ed.")
15
Examen Físico 2 Desvestir el paciente y realizar examen completo
Aliento y olor del paciente ( almendras: cianuro; frutas : alcoholes; Ajo :arsénico; organofosforados: pescado; fósforo: zinc). RsRs y RsCs: arrirtmias, estertores. RsIs: disminuidos o aumentados Evidencia externa de trauma Marcas de pinchazos: fosa antecubital, lengua, tobillos. Bryant, Sean. Management of toxic exposure in children. Emerg Med Clin N Am 21 (2003) Marx, Jhon. Rosen's Emergency Medicine: Concepts and Clinical Practice, Elsevvier. 6th ed.
. RsRs y RsCs: arrirtmias, estertores. RsIs: disminuidos o aumentados. Evidencia externa de trauma. Marcas de pinchazos: fosa antecubital, lengua, tobillos. Bryant, Sean. Management of toxic exposure in children. Emerg Med Clin N Am 21 (2003) Marx, Jhon. Rosen s Emergency Medicine: Concepts and Clinical Practice, Elsevvier. 6th ed.")
16
Examen Neurológico 3 Nivel de conciencia Tamaño y reactividad pupilar
Presencia o ausencia de nistagmus Reflejos Focalización neurológica Bryant, Sean. Management of toxic exposure in children. Emerg Med Clin N Am 21 (2003) Marx, Jhon. Rosen's Emergency Medicine: Concepts and Clinical Practice, Elsevvier. 6th ed.
 Marx, Jhon. Rosen s Emergency Medicine: Concepts and Clinical Practice, Elsevvier. 6th ed.")
17
4 BRADICARDIA TAQUICARDIA Free base: cocaína Anticolinérgicos: antihistamínicos, antipsicóticos, anfetaminas Simpaticomiméticos Teofilina, hormonas tiroideas P Propanolol, fisostigmine Anticolinesterasas Clonidina, BCC Etanol, alcoholes Digital f A A C s E t D

18
5 Hipota hta Cocaína Suplemento tiroideo Simpaticomimético Cafeína C Clonidina Raticidas: arsénico ATC, aminofilina, antiHTA Sedativo, hipnóticos Heroína C R t A s S c H

19
HIPERVENTILACIÓN HIPOVENTILACIÓN
6 HIPERVENTILACIÓN HIPOVENTILACIÓN Paraquat Asa Nervioso agentes Toxinas acidosis metabólica S Sedativo hipnótico Licor Opiodes Wedd: marihuana P L A O N W T

20
7 Colinérgicos Barbitúricos Carbamatos Opioides Clonidina
Nicotina Fenotiazina Fisostigmina Pilocarpina Anticolinérgicos Anfetaminas Cocaína LSD IMAOs Simpaticomiméticos

21
Toxidromes 8 Síndromes clínicos
Reconocimiento de patrones tóxicos o de envenenamiento. Constelación de Síntomas y signos

22
Simpático mimético Sedativo opiáceo Serotoninérgico Anticolinérgico Colinérgico

23
Cocaína, anfetaminas, LSD, descongestionantes
Simpaticomimético Midriasis Taquicardia HTA HiperT° Convulsiones Alucinaciones Casos graves arritmias Cocaína, anfetaminas, LSD, descongestionantes

24
Anticolinérgico Midriasis Taquicardia Hipertensión Íleo
Retención urinaria Piel Seca Fiebre Visión borrosa Psicosis Alucinaciones Mioclonias Coma/Delirio HIPERTA DISMINUCIÓN RUIDOS Antihistamínicos, fenotiazida, escopolamina, atropina, antiparkinsonianos

25
Colinérgico Miosis Cefalea Bradicardia Confusión Salivación
Lagrimeo Diarrea Emesis Diaforesis Broncorrea Inc. Urinaria y fecal Cefalea Confusión Fasciculaciones musculares Calambres Parálisis Flácida Insecticidas Organofosforados o carbamatos, algunos Hongos, Fisostigmina

26
ISRS, fluoxetina, sertralina, paroxetina
Serotoninérgico Taquicardia Midriasis Piel caliente y sudorosa vómito,diarrea Irritabilidad, hiperreflexia Trismos, temblor, mioclonias. ISRS, fluoxetina, sertralina, paroxetina

28
APROXIMACION DIAGNOSTICA

29
Laboratorio de Toxicología
Glucosa, electrolitos, Función renal, Hepática EKG: Medir QT y QRS Pulsoximetría Gases arteriales

30
Laboratorio de Toxicología
Sangre: 10 cc en tubo seco. Orina: La mayor cantidad posible. Contenido gástrico? Solo para cadena de custodia Bryant, Sean. Management of toxic exposure in children. Emerg Med Clin N Am 21 (2003) Tamara, Mc Gregor. Evaluation and management of common chilhood poisonings. American family Physician.Vol 79 Number 5 (2009)
 Tamara, Mc Gregor. Evaluation and management of common chilhood poisonings. American family Physician.Vol 79 Number 5 (2009)")
31
Anión Gap Normal 8- 16 mEq/L.
Timothy B. Erickson. The Approach to the Patient with an Unknown Overdose. Emerg Med Clin N Am 25 (2007) 249–281 Holstege Christopher. Critical care toxicology. Emerg Med Clin N Am 25 (2008)
 249–281. Holstege Christopher. Critical care toxicology. Emerg Med Clin N Am 25 (2008)")
32
Osmolar GAP -15 a + 10 mOsm/ Kg : normal
Incremento presencia de sustancia de bajo peso molecular, osmóticamente activa en el suero Diferencia entre osmolaridad medida y la calculada por laboratorio Holstege Christopher. Critical care toxicology. Emerg Med Clin N Am 25 (2008)
")
33
Rx de Abdomen Timothy B. Erickson. The Approach to the Patient with an Unknown Overdose. Emerg Med Clin N Am 25 (2007) 249–281
 249–281.")
34
REANIMACION Y MANEJO INICIAL

35
DEXTROSA OXíGENO NALOXONA TIAMINA Timothy B. Erickson. The Approach to the Patient with an Unknown Overdose. Emerg Med Clin N Am 25 (2007) 249–281
 249–281.")
36
Cadena de Supervivencia Pediátrica: PREVENCIÓN
B C E Cadena de Supervivencia Pediátrica: PREVENCIÓN Reanimación del paciente Diagnóstico clínico: Toxidrómes Descontaminación Antídoto American Heart Association Guidelines for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care. Circulation. 2010

37
CIRCULACIÓN C Tensión arterial y pulso EKG Líneas venosas
Determinar la presencia de: * Bradicardia/bloqueo AV * Intervalo QRS prolongado * Taquicardia * Arritmias ventriculares Usama A. Hanhan. The Poisoned Child in the Pediatric Intensive Care Unit. Pediatr Clin N Am 55 (2008) 669–686
 669–686.")
38
Evaluación de reflejos protectores. Posición del paciente.
B A VIA AEREA VENTILACION Evaluación de reflejos protectores. Posición del paciente. Limpieza y succión de la vía aérea. ¿Entubación endotraqueal?. GA Asistencia ventilatoria. Oxígeno. * Falla ventilatoria. * Hipoxia * Broncoespasmo. American Heart Association Guidelines for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care. Circulation. 2010

39
DÉFICIT NEUROLÓGICO Estado neurológico Evaluar glicemia
Considerar causas orgánicas * Hipoglicemia * Coma o estupor * Hipotermia o hipertermia * Convulsiones * Agitación

40
Descontaminación DESCONTAMINACIÓN SUPERFICIAL E Piel Ojos Inhalación
DESCONTAMINACIÓN GASTROINTESTINAL Lavado gástrico Carbón activado Irrigación intestinal total

41
MANEJO DEFINITIVO

42
Manejo Inicial Shock : Convulsión: Hipoglucemia sintomática: Naloxona:
10-20 ml/kg de LEV Convulsión: Diazepam 0,3 mg /kg Hipoglucemia sintomática: DAD al % . Tiamina para prevenir la Enc. De Wernicke Glucagón 10 mg IM sin acceso IV Naloxona: 0,1 mg/kg (máx. 2 mg dosis) Tamara, Mc Gregor. Evaluation and management of common chilhood poisonings. American family Physician.Vol 79 Number 5 (2009)
 Tamara, Mc Gregor. Evaluation and management of common chilhood poisonings. American family Physician.Vol 79 Number 5 (2009)")
43
Tamara, Mc Gregor. Evaluation and management of common chilhood poisonings. American family Physician.Vol 79 Number 5 (2009)
")
44
¿Cuál es la indicación de Naloxona?
Se recomienda en pacientes con sobredosis con opiáceos. Se debe soportar ventilatoriamente al paciente antes de su administración. Su uso puede evitar la intubación del paciente. Dosis: 0.1 mg /Kg IV. Hoffman JR. The empiric use of naloxone in patients with altered mental status: a reappraisal. Ann Emerg Med 1991;20:246–52. Gill AM, Cousins A, Nunn AJ, Choonara JA. Opiate-induced respiratory depression in pediatric patients. Ann Pharmacother. 1996; 30: 125–129

45
ANTIDOTOS ESPECIFICOS

46
Mantenimiento 50mg/kg para 4 hr y 100/mg/kg para 12hr
Acetaminofén N-Acetil Cisteina Bolo 150mg/kg/IV Mantenimiento 50mg/kg para 4 hr y 100/mg/kg para 12hr Anticolinérgicos (escopolamina) Salicilato de Fisostigmine 1-2mg/IV en infusión lenta cada 3 a 5 min Benzodiacepina Flumazenil 0,2mg IV en bolo dosis máxima 3mg Carbamatos Atropina 2-4 mg IV en bolo, se repite según respuesta Digoxina Fragmentos de anticuerpo contra digoxina 10 a 20 ampollas IV para situaciones que comprometan la vida Acetaminofén: N acetil cisteina. Metabolizado por el hígado a cisteina que es precursos de glutation, que provee grupos sulfidrilo que penetran hepatocito y se unen al metabolito tóxico. 140mg/kg seguido de 70 mg/kg C 4 horas por 17 dosis Anticolinergico: Antidoto es fisostigmina (amina terciaria) inhibe la colinesterasa. Dosis 0.5 – 2mg/IV en 5 minutos, monitoreo BZP: Flumazenil inhibicion competitiva en los sotios de recptor, y antagonista
 Salicilato de Fisostigmine. 1-2mg/IV en infusión lenta cada 3 a 5 min. Benzodiacepina. Flumazenil. 0,2mg IV en bolo dosis máxima 3mg. Carbamatos. Atropina. 2-4 mg IV en bolo, se repite según respuesta. Digoxina. Fragmentos de anticuerpo contra digoxina. 10 a 20 ampollas IV para situaciones que comprometan la vida. Acetaminofén: N acetil cisteina. Metabolizado por el hígado a cisteina que es precursos de glutation, que provee grupos sulfidrilo que penetran hepatocito y se unen al metabolito tóxico. 140mg/kg seguido de 70 mg/kg C 4 horas por 17 dosis. Anticolinergico: Antidoto es fisostigmina (amina terciaria) inhibe la colinesterasa. Dosis 0.5 – 2mg/IV en 5 minutos, monitoreo. BZP: Flumazenil inhibicion competitiva en los sotios de recptor, y antagonista.")
47
Metanol Etanol 10% 10 cc/kg IV u oral Metilpirazol 15 mg/kg en bolo, puede repetirse Plomo, Arséico, mercurio, antimonio, Bismuto, cobre, hierro, zinc Dimercaprol (si hay encefalopatía) 3 – 5mg/IM o 50 – 75 mg/m2 IM por 10 dias EDTA en 250cc, infuión en : 20-30 mg/k duidos 12 – 20 cada hora Cyanida Amylnitrato, nitrito de sodio, tiosulfato de sodio Calcio antagonistas Calcio al 10% 10 – 20 cc IV en bolo, repetir dosis o infuaión Glucagón 5 – 10mg IV en bolo seguido de5-10/horas en infiusión.
 3 – 5mg/IM o. 50 – 75 mg/m2 IM por 10 dias. EDTA en 250cc, infuión en : mg/k duidos 12 – 20 cada hora. Cyanida. Amylnitrato, nitrito de sodio, tiosulfato de sodio. Calcio antagonistas. Calcio al 10% 10 – 20 cc IV en bolo, repetir dosis o infuaión. Glucagón. 5 – 10mg IV en bolo seguido de5-10/horas en infiusión.")
48
2 – 4mg/IV en bolo repetir dosis
Opioides Naloxona 0.4 – 2 mg IV Organosfosforados Atropina Pralidoxina: 2 – 4mg/IV en bolo repetir dosis 1 a 2 gr dia Antidepresivos tricíclicos Bicarbonato de Sodio 1 – 2 mEq IV en bolo Hierro Deferoxamina (Desferal) Infusión IV 15 mg/kg hora Raticidas: vitamina K
 Infusión IV 15 mg/kg hora. Raticidas: vitamina K.")
49
DESCONTAMINACION

50
Descontaminación Gastrointestinal
Avila Albert; Moreno Atilio. Medidas generales en el servicio de urgencias para el paciente pediátrico intoxicado. Trabajo de promoción. Medicina de Urgencias. Pontificia Universidad Javeriana ( pendiente publicación) Bryant, Sean. Management of toxic exposure in children. Emerg Med Clin N Am 21 (2003) Tamara, Mc Gregor. Evaluation and management of common chilhood poisonings. American family Physician.Vol 79 Number 5 (2009) Marx, Jhon. Rosen's Emergency Medicine: Concepts and Clinical Practice, Elsevvier. 6th ed.
 Bryant, Sean. Management of toxic exposure in children. Emerg Med Clin N Am 21 (2003) Tamara, Mc Gregor. Evaluation and management of common chilhood poisonings. American family Physician.Vol 79 Number 5 (2009) Marx, Jhon. Rosen s Emergency Medicine: Concepts and Clinical Practice, Elsevvier. 6th ed.")
51
a. Inducción del vómito El Jarabe de Ipecacuana NO se recomienda para uso rutinario en pacientes intoxicados. American Academy of Clinical Toxicology and European Association of Poisons Control Centers and Clinical Toxicologists. Position statement: ipecac syrup. J Toxicol Clin Toxicol. 1997;35:699–709.

52
b. Lavado Gástrico Indicaciones
Sustancias que no tengan capacidad de fijarse al carbón activado, no corrosivo, no más de 1 hora de ingestión.

53
Contraindicaciones .Depresión del estado de conciencia .No protección de la vía aérea .Sustancias corrosivas o hidrocarburos .Perforación o hemorragia gastrointestinal

54
Técnica Perforación de esófago o estómago
Verificar posición de la sonda SNG: 22 – 28 Fr en niños- 36 Fr en adolescentes SSN 0,9% 10 ml/kg Volumen de retorno igual al administrado. Líquido claro Perforación de esófago o estómago Broncoaspiración. Estimulación vagal

55
c. Carbón Activado Estrategia más recomendada
Funciona como un efectivo absorbente Ideal primeros 60 min. Máx 2 horas Dosis 0,5- 2 mg/Kg ( Máx 50 gr)
")
56
Indicaciones Exposición por vía oral a sustancias tóxicas, en cantidades, tóxicas Menos de una hora de la ingestión No deterioro del estado de conciencia. Ante cualquier evidencia de deterioro neurológico o la aparición de convulsiones, la administración de carbón activado debe hacerse previa intubación orotraqueal.

57
Timothy B. Erickson. The Approach to the Patient with an Unknown Overdose. Emerg Med Clin N Am 25 (2007) 249–281
 249–281.")
58
Contraindicaciones Carece de utilidad o está contraindicado en la exposición a las siguientes sustancias: alcoholes, hidrocarburos, ácidos y álcalis, hierro, litio.

59
Técnica Añadir 8 partes de agua a la cantidad seleccionada en forma de polvo. Paso por sonda nasogástrica u orogástrica.

60
d. Irrigación Intestinal
INDICACIONES: Agentes no bien absorbidos por carbón activado Restos de pinturas plomadas Tabletas de hierro Pilas Transportadores de drogas “ mulas” Calcio antagonistas Ingestión de sustancia de liberación prolongada o cubierta entérica Paquetes o condones

61
DOSIS: 25 -50 cc/kg/ h máx 2 lt/h hasta que el líquido sea claro
DOSIS: cc/kg/ h máx 2 lt/h hasta que el líquido sea claro. Su efecto inicia en 1 hora. Polietilenglicol: Isotónico, no absorbible, No alteraciones hidroelectrolíticas. Se disuelve en 1 lt de agua y se pasa por SNG u oral.

62
e. Medidas de Excreción Alcalinización de la orina :pH mayor o igual a 7,5 Promueve eliminación de ácidos débiles Bicarbonato de Sodio 1-2 meq/Kg. ( 15 ampollas en 850 cc de DAD 5%. Pasar a 2-3 cc/kg/h). Antidepresivos tricíclicos ASA Fenobarbital Herbicidas
. Antidepresivos tricíclicos. ASA. Fenobarbital. Herbicidas.")
63
Timothy B. Erickson. The Approach to the Patient with an Unknown Overdose. Emerg Med Clin N Am 25 (2007) 249–281
 249–281.")
64
INTOXICACIONES ESPECIFICAS

65
INTOXICACION POR ACETAMINOFEN

66
INTOXICACION POR ACETAMINOFEN
Principal causa de intoxicaciones Absorción en 2 horas Dosis terapéuticas: mg/kg/dosis Dosis toxicas: mg/kg (vol: peso*5) Intoxicación a dosis mas bajas si hay niveles bajos de glutation, desnutrición, o situaciones que sean inductoras de enzimas: uso de anticonvulsivantes, isoniazida La dosis terapéutica del acetaminofén es de mg/kg dosis o mg/kg/día. En general se deben utilizar máximo 2gr al día en niños y 4gr al día en adultos. Se considera que una dosis mayor a mg/kg en niños o 6 -7gr en adultos es potencialmente tóxico agudo. La dosis letal del acetaminofén es de 13-25gr. Se debe solicitar a los familiares los frascos empaques de las tabletas y mirar la presentación para calcular según el número de tabletas ingeridas los mg/kg de peso que pudo consumir el paciente. Se considera que un paciente puede presentar toxicidad crónica si ingiere más de 4 g/día especialmente en pacientes con bajos niveles de glutatión o inducción de enzimas hepáticas como en alcoholismo, desnutrición y uso de medicamentos como anticonvulsivantes. En niños dosis de mg/Kg/día por 2-8 días se consideran como posiblemente tóxicas crónicas.
 Intoxicación a dosis mas bajas si hay niveles bajos de glutation, desnutrición, o situaciones que sean inductoras de enzimas: uso de anticonvulsivantes, isoniazida. La dosis terapéutica del acetaminofén es de mg/kg dosis o mg/kg/día. En general se deben utilizar máximo 2gr al día en niños y 4gr al día en adultos. Se considera que una dosis mayor a mg/kg en niños o 6 -7gr en adultos es potencialmente tóxico agudo. La dosis letal del acetaminofén es de 13-25gr. Se debe solicitar a los familiares los frascos empaques de las tabletas y mirar la presentación para calcular según el número de tabletas ingeridas los mg/kg de peso que pudo consumir el paciente. Se considera que un paciente puede presentar toxicidad crónica si ingiere más de 4 g/día especialmente en pacientes con bajos niveles de glutatión o inducción de enzimas hepáticas como en alcoholismo, desnutrición y uso de medicamentos como anticonvulsivantes. En niños dosis de mg/Kg/día por 2-8 días se consideran como posiblemente tóxicas crónicas.")
67
Metabolismo 90% hepático 3 rutas metabólicas diferentes:
70% conjugado con glucurónido 20%conjugado con sulfato oxidación con cpy450 mas conjugación con glutation. Approximately 5% of a therapeutic dose is metabolized by cytochrome P450 2E1 to the electrophile N-acetyl-p-benzoquinone imine (NAPQI).8 NAPQI is extremely toxic to the liver, possibly as a result of covalent binding to proteins and nucleic acids.9 However, NAPQI is rapidly detoxified by interaction with glutathione to form cysteine and mercapturic acid conjugates.10
.8. NAPQI is extremely toxic to the liver, possibly as. a result of covalent binding to proteins and nucleic. acids.9 However, NAPQI is rapidly detoxified. by interaction with glutathione to form cysteine. and mercapturic acid conjugates.10.")
68
Fisiopatología 3-5% : N – acetil- para-benzoquinoneimina.
Altamente hepato y nefrotoxica Glutation: molécula hidrosoluble, menos activa Eliminada por la orina Altas dosis: mayor producción de NAPQI excede la capacidad del glutatión Normalmente se metaboliza el 3-5% en N – acetil- para-benzoquinoneimina. Altamente hepato y nefrotoxica, se une al glutation y forma una molecula hidrosoluble capaz de ser eliminada por la orina, en dosis toxicas la produccion de NAPQI es mayor que la capacidad del glutation La mayoria del metabolisno se produce en la zona 3 del acino hepatico, lo que hace que esta zona sea la primera afectada. Y altas dosis esta se´puede producir necrosis de las zonas I y II tambien. FISIOPATOLOGÍA Este analgésico derivado de la fenacetina, se absorbe muy bien por vía oral y se logran concentraciones máximas a los 30–60 minutos; se une poco a las proteínas plasmáticas. Es metabolizado a nivel hepático por el citocromo p-450 a metabolitos inactivos. Se produce un 3–5% de un metabolito menor muy activo que es la N – acetil- para-benzoquinoneimina (NAPQI), responsable de la lesión hepática y renal. Menos de 5% se excreta en la orina sin cambios. Normalmente la NAPQI es rápidamente eliminada por el glutatión en el hígado. Sin embargo, en sobredosis, la producción del metabolito tóxico excede la capacidad del glutatión y el metabolito reacciona directamente con macromoléculas hepáticas, lo que produce daño hepático. La sobredosis durante el embarazo ha sido asociada con muerte fetal y aborto espontáneo. DOSIS TÓXICA • Adultos 6 –7 g. • Dosis letal g. • Niños: En ingestiones agudas, 150 – 200 mg/kg. El riesgo de hepatotoxicidad se incrementa si el paciente es alcohólico crónico o si toma otros medicamentos que incrementen la actividad del sistema de citocromo- oxidasas, como lanticonvulsivantes e isoniazida. En la desnutrición se disminuye el glutation corporal, por lo cual ante la sobredosis de acetaminofén no es posible eliminar el NAPQI, metabolito sumamente tóxico. The oxidation of acetaminophen by CYP450 subfamilies, predominantly CYP2E1 [4] [5] results in the formation of the highly reactive electrophile, N-acetyl-p-benzoquinoneimine (NAPQI).[3] NAPQI combines rapidly with glutathione and other thiol-containing compounds, forming nontoxic conjugates, which are eliminated in urine. When NAPQI formation exceeds glutathione supply, free NAPQI binds to hepatocyte intracellular proteins, causing toxicity. Inducers (e.g., ethanol, isoniazid, anticonvulsants) and inhibitors (e.g., cimetidine) of CYP450 enzymes may affect the NAPQI formation, but their clinical significance is controversial. Renal injury may occur with or without hepatic injury[6] and may be mediated by the presence of CYP450 enzyme species [7] [8] and the activation of prostaglandin synthase within the kidneys. [9] [10] Most oxidative metabolism is concentrated in hepatic zone III, which is affected most via acetaminophen toxicity. In cases of severe toxicity, necrosis can extend into zones I and II, destroying the entire liver parenchyma. The sequelae of severe acetaminophen toxicity are those of fulminant liver failure, rather than direct acetaminophen effects, and the pathophysiology of these complex multisystem problems is well described (see Chapter 89 ).[
, responsable de la lesión hepática y. renal. Menos de 5% se excreta en la orina sin. cambios. Normalmente la NAPQI es rápidamente eliminada. por el glutatión en el hígado. Sin embargo, en sobredosis, la producción del metabolito. tóxico excede la capacidad del glutatión. y el metabolito reacciona directamente con. macromoléculas hepáticas, lo que produce. daño hepático. La sobredosis durante el embarazo. ha sido asociada con muerte fetal. y aborto espontáneo. DOSIS TÓXICA. • Adultos 6 –7 g. • Dosis letal g. • Niños: En ingestiones agudas, 150 – 200. mg/kg. El riesgo de hepatotoxicidad se incrementa si. el paciente es alcohólico crónico o si toma. otros medicamentos que incrementen la actividad. del sistema de citocromo- oxidasas, como lanticonvulsivantes e isoniazida. En la. desnutrición se disminuye el glutation corporal, por lo cual ante la sobredosis de acetaminofén. no es posible eliminar el NAPQI, metabolito. sumamente tóxico. The oxidation of acetaminophen by CYP450 subfamilies, predominantly CYP2E1 [4] [5] results in the formation of the highly reactive electrophile, N-acetyl-p-benzoquinoneimine (NAPQI).[3] NAPQI combines rapidly with glutathione and other thiol-containing compounds, forming nontoxic conjugates, which are eliminated in urine. When NAPQI formation exceeds glutathione supply, free NAPQI binds to hepatocyte intracellular proteins, causing toxicity. Inducers (e.g., ethanol, isoniazid, anticonvulsants) and inhibitors (e.g., cimetidine) of CYP450 enzymes may affect the NAPQI formation, but their clinical significance is controversial. Renal injury may occur with or without hepatic injury[6] and may be mediated by the presence of CYP450 enzyme species [7] [8] and the activation of prostaglandin synthase within the kidneys. [9] [10] Most oxidative metabolism is concentrated in hepatic zone III, which is affected most via acetaminophen toxicity. In cases of severe toxicity, necrosis can extend into zones I and II, destroying the entire liver parenchyma. The sequelae of severe acetaminophen toxicity are those of fulminant liver failure, rather than direct acetaminophen effects, and the pathophysiology of these complex multisystem problems is well described (see Chapter 89 ).[")
69
La mayoría del metabolismo se produce en la zona 3 del acino hepático, lo que hace que esta zona sea la primera afectada. Y altas dosis esta se puede producir necrosis de las zonas I y II también.

70
Fases de la intoxicación
tiempo características 1 Primeras 24 horas Síntomas inespecíficos, nauseas, vomito, diaforesis, pueden resolver 2 24-72 horas Lesión hepática, dolor en HCD y epigastrio, nauseas, vomito, elevación de enzimas, hepáticas, hepatomegalia, oliguria, hiperbilirrubinemia, prolongación de los tiempos de coagulación. 3 72-96 horas Necrosis hepática, falla hepática, encefalopatía, acidosis metabólica hipoglucemia falla renal aguda, SFOM, edema cerebral, muerte 4 7-14 días Resolución de la disfunción hepática SIN DISFUNCION HEPATICA CRONICA. CLINICAL FEATURES The progression of acetaminophen-induced hepatic injury occurs in four stages. Stage 1 is the preinjury period, usually the first 24 hours after ingestion. Nonspecific symptoms, including nausea, vomiting, anorexia, diaphoresis, and malaise, are common in the first 8 hours after ingestion, usually resolving thereafter. Patients may be asymptomatic during this stage. Stage 2 is the onset of liver injury, usually 24 hours after ingestion but possibly 12 to 36 hours after overdose. In extraordinary cases, liver injury is evident as early as 8 hours after ingestion. Signs and symptoms of hepatic injury include nausea, vomiting, and right upper quadrant and epigastric pain or tenderness. Stage 3 is maximum liver injury, usually 3 to 4 days after ingestion, with a wide variety of clinical manifestations, depending on severity of the injury. Signs and symptoms of hepatic injury can persist or progress. Fulminant hepatic failure can develop during this stage, with encephalopathy, coma, and clinical evidence of coagulopathy. Patients with hepatic failure sometimes develop hypoglycemia and metabolic acidosis. Death with hepatic failure can occur from hemorrhage, adult respiratory distress syndrome, sepsis, multiorgan failure, or cerebral edema. [2] [15] The risk of renal injury increases with severity of hepatic injury, occurring in less than 2% of patients without hepatotoxicity and in 25% of patients with severe hepatotoxicity. [16] [17] [18] Stage 4 is the recovery period. Hepatic enzymes return to baseline by 5 to 7 days but may take longer with severe hepatic injury. Complete histologic resolution of hepatic insult can take months. Regeneration of the liver is complete, without chronic hepatic dysfunction.[19]

71
DX Y TTO Sospecha clínica
Concentración de acetaminofén en sangre, primeras 24 horas Enzimas hepáticas, electrolitos, tiempos de coagulación, BUN, creatinina, glicemia, bilirrubinas, gases arterialeS Tratar si: Siempre si la dosis es tóxica Alteración de pruebas de función hepática Concentración en línea de tratamiento Concentración mayor a 10mcg/dl si no se conoce la hora de ingestión

72
Metas de tratamiento Limitar la absorción: se podría hacer lavado gástrico si la consulta es muy rápida, carbón activado Iniciar antídoto lo mas rápido posible Control de los síntomas Soportes que requiera

73
Antídoto N-ACETILCISTEÍNA
DOSIS DE CARGA DE 140 MG/KG, MANTENIMIENTO: 70 MG/KG CADA 4 HORAS-17 DOSIS. Máximo beneficio las primeras 10 horas Control paraclinico y evolución del paciente definen si se debe continuar por mas tiempo la NAC Asociar a antiemético para no perder dosis administrada, si vomita en la 1 hora después de la dosis se debe repetir la dosis Acetylcysteine (also known as N-acetylcysteine) prevents hepatic injury primarily by restoring hepatic glutathione (Fig. 1B).10 In addition, in patients with acetaminophen-induced liver failure, acetylcysteine improves hemodynamics and oxygen use,12 increases clearance of indocyanine green (a measure of hepatic clearance),13 and decreases cerebral edema.14 The exact mechanism of these effects is not clear, but it may involve scavenging of free radicals or changes in hepatic blood flow.12,1
 prevents hepatic injury primarily by restoring. hepatic glutathione (Fig. 1B).10 In addition, in patients. with acetaminophen-induced liver failure, acetylcysteine improves hemodynamics and oxygen. use,12 increases clearance of indocyanine. green (a measure of hepatic clearance),13 and. decreases cerebral edema.14 The exact mechanism. of these effects is not clear, but it may involve. scavenging of free radicals or changes in hepatic. blood flow.12,1.")
75
Intoxicación por hierro
Prevención y tratamiento de Anemia por deficiencia de Hierro en todas las edades Primeros reportes de intoxicación fueron a mitad del siglo XX 1997 la FDA advierte sobre el riesgo de intoxicación

76
Hierro Por ingestión de multivitamínicos.
Intoxicaciones mas graves por medicamentos para adultos Niveles normales: mcg/dl Unido a transferrina: mcg/dl, y cuando se satura al 100%, empieza a circular Fe libre se oxida y puede generar daño celular 80% se utiliza en médula ósea y 20 % para deposito

77
PRESENTACION -Sultafo ferroso -gotas 125 mg/ml , equivalente a 25 mg de hierro elemental - grag 100mg - jarabe 200mg/5ml

78
Dosis tóxicas Síntomas moderados 20-60 mg/kg
Síntomas graves: mas de 60 mg/kg Dosis letal en el 50% de las personas: mg/kg 130 mg/kg Riesgo de muerte en niños

80
MECANISMOS DE TOXICIDAD
Lesión caustica directa a la mucosa del TGI

81
Mecanismos de toxicidad
. Fe pasa al interior de la cel y se acumula en crestas mitocondriales, desacopla la fosforilacion oxidativa, lesion y muerte cel y lib de radicales libres, prod peroxidacion de membranas. Efectos tóxicos ocurren en dosis de 20 mg /k Dosis letal 60 mg/k Mecanismo de lesión: Desacople de la Fosforilación oxidativa Acidosis Metabólica : 3Fe3* + H2O =Fe 2* + H + Efecto corrosivo directo Formación de radicales libres por ciclo de oxidoreducción Desacople de la fosforilacion oxidativa y no producción de ATP

82
Mecanismo de toxicidad
Lesión caustica directa en mucosa TGI: vómito diarrea, hemorragia y dolor abdominal, perforación, peritonitis. Alteración del metabolismo intracelular: principalmente corazón, hígado y SNC por liberación de radicales libres Vasodilatación sistémica: shock distributivo. Aumentan la permeabilidad capilar: shock hipovolémico Acidosis Metabólica : 3Fe3* + H2O =Fe 2* + H

83
Diagnóstico Sospecha Rx. abdomen Fe en sangre las primeras 24 horas
300 mcg/dl a 500 mcg/dl toxicidad gastrointestinal 500 mcg/dl a 1000 mcg/dl toxicidad sistémica severa Mayor de 1000 mcg/dl toxicidad letal Glicemia Hemograma Gases arteriales Prueba de provocación con desferoxamina: quelación del Fe que produce orina vino rosado. Fe serico: no confiable pq se deposita en higado Prueba de provocacion con desferoxamina: quelacion del Fe que prod orina vino rosado. Rx abd

84
Cuadro clínico Fase I: vómito durante los prim 80 min y posteriormente diarrea, hemorragia, shock y muerte. Fase II: recuperación aparente por +/-24 horas, AUMENTO DE LA ACIDOSIS Fase III: reaparición de HVD, deterioro del nivel de conciencia, shock hipovolémico y distributivo, acidosis metabólica, leucocitosis, coagulopatía (temprana por alt. de trombina, tardía por falla hepática) Fase IV: 3-5 días post ingestión se produce falla hepática aguda: ictericia, coma hepático, elevación de transaminasas, elevación de amoniaco, hipoglucemia, coagulopatía, necrosis periportal, necrosis grasa de cel. de kupffer, miocardio degeneración grasa Fase V: tardía obstrucción intestinal por lesion inicial en píloro e intestino prox. ESTADIO I 30 min a 2 horas Gastrointestinal Náuseas, vómito, dolor abdominal y diarrea Hematemesis, melenas o hematoquezia Ausencia de vómito, en las primeras 6 horas excluye toxicidad grave ESTADIO II Latente 6 a 24 horas posingesta Mejoría de sintomatología gastrointestinal Empeoramiento de Acidosis metabólica Pacientes No tienen toxicidad grave Signos vitales alterados Estado mental normal Estado ácido-base normal Tolerancia a la vía oral ESTADIO III Sistémica o de Primeras horas después de dosis masivas 12 a 24 h después de ingestión moderada Shock por hipovolemia y vasodilatación con disminución de la perfusión sanguínea Coagulopatía Temprana por alteración de función de la trombina Tardía por falla hepática Letargía, convulsiones y coma ESTADIO IV Falla hepática Ocurre 2 a 3 días Hierro es captado por sistema reticuloendotelial donde causa daño oxidativo Necrosis periportal hemorrágica y degeneración grasa de las células de Kupffer o párenquimatosas. Miocardio puede presentar degeneración grasa. ESTADIO V Secuelas Obstrucción gastrointestinal secundario al efecto corrosivo de este inicialmente Ocurre 2 a 8 semanas posterior a la intoxicación Riesgo de infección por Yersinia enterocolitica: Dolor abdominal Fiebre Diarrea
 Fase IV: 3-5 días post ingestión se produce falla hepática aguda: ictericia, coma hepático, elevación de transaminasas, elevación de amoniaco, hipoglucemia, coagulopatía, necrosis periportal, necrosis grasa de cel. de kupffer, miocardio degeneración grasa. Fase V: tardía obstrucción intestinal por lesion inicial en píloro e intestino prox. ESTADIO I. 30 min a 2 horas. Gastrointestinal. Náuseas, vómito, dolor abdominal y diarrea. Hematemesis, melenas o hematoquezia. Ausencia de vómito, en las primeras 6 horas excluye toxicidad grave. ESTADIO II. Latente. 6 a 24 horas posingesta. Mejoría de sintomatología gastrointestinal. Empeoramiento de Acidosis metabólica. Pacientes No tienen toxicidad grave. Signos vitales alterados. Estado mental normal. Estado ácido-base normal. Tolerancia a la vía oral. ESTADIO III. Sistémica o de. Primeras horas después de dosis masivas. 12 a 24 h después de ingestión moderada. Shock por hipovolemia y vasodilatación con disminución de la perfusión sanguínea. Coagulopatía. Temprana por alteración de función de la trombina. Tardía por falla hepática. Letargía, convulsiones y coma. ESTADIO IV. Falla hepática. Ocurre 2 a 3 días. Hierro es captado por sistema reticuloendotelial donde causa daño oxidativo. Necrosis periportal hemorrágica y degeneración grasa de las células de Kupffer o párenquimatosas. Miocardio puede presentar degeneración grasa. ESTADIO V. Secuelas. Obstrucción gastrointestinal secundario al efecto corrosivo de este inicialmente. Ocurre 2 a 8 semanas posterior a la intoxicación. Riesgo de infección por Yersinia enterocolitica: Dolor abdominal. Fiebre. Diarrea.")
85
Tratamiento Estabilización y prevención de shock:
Asegurar la vía aérea Aumentar la Fio2 LEV: bolos de cc/kg

86
Tratamiento Limitar absorción y daño TGI: Hidroxido de magnesio
Lavado gástrico con Agua o solución salina a 15 ml/k hasta que salga limpio y claro Qx: si bezoar (hasta gastrectomia) Polietinlenglico-electrolitos (Nulitely ): cc/kg, continuo hasta que el contenido rectal sea claro, y no se vean en Rx, no en obstrucción o íleo
 Polietinlenglico-electrolitos (Nulitely ): cc/kg, continuo hasta que el contenido rectal sea claro, y no se vean en Rx, no en obstrucción o íleo.")
87
Tratamiento Considerar transfusión en niveles de 1000mcg/dl o mas
Desferoxamina: 100 mg quelan 9,35 mg Fe, dar en infusion a 15 mg/kg/h, se puede dializar. DESFEROXAMINA Quelante específico de Hierro Extraído de Streptomyces pilosus. Con ion férrico forma complejo llamado ferrosamina Complejo soluble que se elimina por riñón Quela el Hierro libre o el unido a trasferrina Acidosis metabólica Vomito que no cede a medicamentos Síntomas de tipo: Letargía, hipotensión sangrado gastrointestinal o signos de Shock con niveles de 150 mcg/dl Niveles por encima de 500 mcg/dl Rayos X de abdomen con presencia de tabletas Infusión intravenosa dosis crecientes desde 15 a 30 mg/k/h por 24 horas No dar más de 6 g/d Suspender cuando: Asintomático Níveles por debajo de 150 mcg/dl Coloración de la orina normal No se observen las tabletas en la placa de abdomen Eventos adversos: Síndrome de distrés respiratorio del adulto cuando infusiones son suériores a 24 h. O cuando dosis se incrementa a más de 6 g/día Sepsis por Yersinia enterocolitica, Zygomycetes, Aeromona Hydrophilia que está caracterizada por dolor abdominal, fiebre y diarrea

89
ORGANOFOSFORADOS

90
Absorción por piel, mucosas y pulmones.
Muy liposolubles Absorción por piel, mucosas y pulmones. Metabolismo hepático (oxidación) con metabolitos mas tóxicos Excreción renal Metabolitos se pueden almacenar en t. adiposo, riñón, hígado y glándulas salivares Estas sustancias se metabolizan a nivel hepatico mediante distintos procesos quimicos, que en oportunidades aumentan la actividad toxica del compuesto, como es el caso del Paration el cual sufre un proceso de oxidacion, formando el compuesto mas toxico Paraoxon. En el caso de fosforados organicos que poseen en su estructura grupos paranitrofenilos, es posible encontrar en la orina de los pacientes, metabolitos como el paranitrofenol. Sus metabolitos pueden almacenarse principalmente en los tejidos adiposos, riñón, higado y glandulas salivares; su excrecion se realiza por via renal en forma relativamente rapida.
 con metabolitos mas tóxicos. Excreción renal. Metabolitos se pueden almacenar en t. adiposo, riñón, hígado y glándulas salivares. Estas sustancias se metabolizan a nivel hepatico. mediante distintos procesos quimicos, que. en oportunidades aumentan la actividad toxica. del compuesto, como es el caso del Paration el. cual sufre un proceso de oxidacion, formando. el compuesto mas toxico Paraoxon. En el caso. de fosforados organicos que poseen en su. estructura grupos paranitrofenilos, es posible. encontrar en la orina de los pacientes, metabolitos. como el paranitrofenol. Sus metabolitos pueden almacenarse principalmente. en los tejidos adiposos, riñón, higado y. glandulas salivares; su excrecion se realiza por. via renal en forma relativamente rapida.")
91
Mecanismo de toxicidad
Se une a la acetilcolinesterasa, fosforilandola unión irreversible Se acumula acetilcolina en la hendidura sináptica o en la placa neuromuscular Sd. colinérgico Los fosforados organicos y carbamatos tienen como accion principal la inhibicion de la enzima acetil-colinesterasa, tanto la colinesterasa eritrocitica o verdadera como la plasmatica o pseudolinesterasa. Los organofosforados actuan por fosforilizacion enzimatica originando una union muy estable que se considera “irreversible”, mientras que los carbamatos actuan por Carbamilacion acumulacion de acetilcolina en la hendidura sinaptica y estimulando excesivamente el SNC, los receptores muscarinicos de las celulas efectoras parasimpaticas, los receptores nicotinicos presentes en la placa neuromuscular y en los ganglios autonomos, traducido clinicamente en un sindrome colinergico.

94
Cuadro clínico Dosis tóxica:
Depende de la potencia del organofosforado o carbamato y de muchos otros factores como la via y el tiempo de exposicion. Es importante conocer la categoria toxicologica del compuesto involucrado para determinar junto con la cantidad ingerida o absorbida por las diferentes vias, la severidad del cuadro clinico y, por lo tanto, tomar las medidas terapeuticas adecuadas. Manifestaciones clínicas: Los fosforados organicos y los carbamatos inhiben las colinesterasas y producen una acumulacion de acetilcolina, produciendo alteraciones en la transmision colinergica de la sinapsis. Los signos y sintomas de este tipo de intoxicacion pueden presentarse dentro de pocos minutos hasta 1 a 2 horas posteriores a la exposicion. Se presenta un deterioro progresivo estableciendose el cuadro

96
Sd intermedio/recaída del 3 día
Aparición horas post exposición Parálisis muscular proximal: pares cran, nuca, musc. respiratorios Teorías: pasa el efecto de la atropina, se reinicia el peristaltismo y hay absorción del toxico, reabsorción de toxico eliminado (sudor) , liberación del toxico almacenado. Alt. card: bloqueo a-v, cambios en ST, prolong. de QT, arritmias ventriculares, Alt pulm: secreción bronquial, bronco espasmo, depresión respiratoria RIESGO DE FALLA RESP Y MUERTE • Síndrome Intermedio: En 1974 Wadia describe un nuevo sindrome, el intermedio, que aparece entre las 24 y 96 horas de iniciada la intoxicacion y que se caracteriza por una paralisis proximal que involucra pares craneanos, musculos flexores de nuca y musculos de la respiracion, originando gran dificultad respiratoria y llevando a la muerte al paciente si no se proporciona soporte ventilatorio. Se han enunciado como posibles responsables el fention, dimetoato, monocrotofos, metamidofos. Se ha descrito como “recaida del tercer dia” y se cree que ello se debe a varios factores: La liposolubilidad del fosforado que le permite almacenarse por algunas horas o dias en el tejido graso del paciente. La prolongada sobre-estimulacion de la acetilcolina en los receptores nicotinicos postsinapticos de la union neuromuscular que causa dano de receptores. La persistencia del toxico a cualquier nivel de organismo primordialmente a nivel del tracto digestivo (especialmente intestinal), tracto que por la “atropinizacion” se somete a paralisis y que coincidencialmente en el tiempo en que se presenta la “recaida” (tercer dia) coincide con la reduccion de la atropinizacion y el reinicio del peristaltismo intestinal, con lo cual se facilita que el material intestinal se ponga en contacto con nuevas partes de mucosa y se produce “reintoxicacion”. La medicacion adecuada y oportuna, y sobre todo la “exhaustiva descontaminacion” especialmente de intestino, de piel, cuero cabelludo, unas, conducto auditivo externo, disminucion de la reabsorcion de metabolitos excretados por sudor o secreciones que persisten en contacto con el paciente, permiten que la incidencia de esta complicacion disminuya. Algunos efectos cardiacos incluyen bloqueo A-V, cambios en el segmento ST, prolongacion del QT y arritmias ventriculares. La hipersecrecion bronquial, broncoconstriccion y depresion respiratoria sobreviene en casos severos lo que puede llevar a falla respiratoria y muerte.
 , liberación del toxico almacenado. Alt. card: bloqueo a-v, cambios en ST, prolong. de QT, arritmias ventriculares, Alt pulm: secreción bronquial, bronco espasmo, depresión respiratoria. RIESGO DE FALLA RESP Y MUERTE. • Síndrome Intermedio: En 1974 Wadia describe. un nuevo sindrome, el intermedio, que aparece entre las 24 y 96 horas de iniciada. la intoxicacion y que se caracteriza. por una paralisis proximal que involucra pares. craneanos, musculos flexores de nuca. y musculos de la respiracion, originando. gran dificultad respiratoria y llevando a la. muerte al paciente si no se proporciona soporte. ventilatorio. Se han enunciado como. posibles responsables el fention, dimetoato, monocrotofos, metamidofos. Se ha descrito. como recaida del tercer dia y se cree. que ello se debe a varios factores: La liposolubilidad del fosforado que le permite. almacenarse por algunas horas o dias. en el tejido graso del paciente. La prolongada sobre-estimulacion de la. acetilcolina en los receptores nicotinicos. postsinapticos de la union neuromuscular. que causa dano de receptores. La persistencia del toxico a cualquier nivel. de organismo primordialmente a nivel del. tracto digestivo (especialmente intestinal), tracto que por la atropinizacion se somete. a paralisis y que coincidencialmente en. el tiempo en que se presenta la recaida (tercer dia) coincide con la reduccion de. la atropinizacion y el reinicio del peristaltismo. intestinal, con lo cual se facilita que. el material intestinal se ponga en contacto. con nuevas partes de mucosa y se produce. reintoxicacion . La medicacion adecuada y. oportuna, y sobre todo la exhaustiva descontaminacion especialmente de intestino, de piel, cuero cabelludo, unas, conducto. auditivo externo, disminucion de la. reabsorcion de metabolitos excretados por. sudor o secreciones que persisten en contacto. con el paciente, permiten que la incidencia. de esta complicacion disminuya. Algunos efectos cardiacos incluyen bloqueo A-V, cambios en el segmento ST, prolongacion del QT. y arritmias ventriculares. La hipersecrecion bronquial, broncoconstriccion y depresion respiratoria. sobreviene en casos severos lo que puede llevar a. falla respiratoria y muerte.")
98
Cuantificación de la inhibición
aguda de la actividad de la enzima acetil colinesterasa: Su determinacion es muy importante en casos de intoxicaciones agudas. Esta prueba nos permite saber si el individuo ha estado en contacto con plaguicidas organofosforados y/o carbamatos y a su vez la intensidad de impacto que ha ejercido sobre el paciente en estudio. Se basa esencialmente en el metodo descrito por Rappaport et al, que mide el acido acetico producido por la hidrolisis enzimatica de acetilcolina. Llevando a cabo la reaccion en presencia de un sustrato tal como el cloruro de acetilcolina, el acido acetico produce disminucion de pH, que es proporcional a la actividad de la colinesterasa presente. El metodo de Michel utiliza un potenciometro para medir la cantidad de acido, segun el cambio de pH producido por la accion de la enzima en una solucion tampon estandar durante un tiempo determinado. La actividad enzimatica se expresa en deltas de pH/hora. El resultado normal es de Uds. Δ pH/hora. La actividad enzimatica varia entre personas; sin embargo se considera significativa una disminucion de mas del 25% de la actividad tomando como referencia el limite inferior de normalidad.

99
Metas de tratamiento Descontaminación Soporte básico
Revertir el exceso de acetilcolina Revertir la unión con la acetilcolinesterasa

100
TRATAMIENTO Tratamiento a. General:
CBA Aumento de secreciones, o2 Quitar ropa contaminada, limpiar bien al pte. agua+HCO3 Na Lavado gástrico mas carbón activado Tratamiento a. General: • Mantenimiento de via aerea con limpieza y aspiracion de secreciones. Oxigenoterapia y observacion permanentemente de la actividad de los musculos respiratorios ya que pueden presentar falla respiratoria aguda. En casos severos se requiere intubacion orotraqueal y ventilacion asistida. • Remover la ropa contaminada y realizar bano con agua y jabon en las zonas expuestas. Sera preferible una segunda limpieza con agua alcalinizada (agua mas bicarbonato de sodio en polvo) si la intoxicacion fue por via dermica ya que el medio alcalino hidroliza el toxico. • No inducir vomito por el riesgo de broncoaspiracion. Ademas las presentaciones liquidas de los plaguicidas muy frecuentemente contienen hidrocarburos tipo kerosene que aumenta el riesgo de producir neumonitis quimica durante la emesis. • Lavado gastrico con abundante suero fisiologico o solucion salina, si la ingesta fue hace menos de 1 hora y protegiendo la via aerea en caso de que el paciente tenga disminucion del estado de conciencia. Administrar carbon activado 1g/kg de peso cada 8 horas para adsorber o atrapar el plaguicida y evitar su absorcion. • La administracion de catartico salino no es recomendada ya que puede exacerbar la gastroenteritis causada por los organofosforados o carbamatos. Ademas el cuadro colinergico a su vez se acompana de diarrea. • Control y manejo de equilibrio acido-basico del paciente. Administrar bicarbonato de sodio segun requerimientos observados en los gases arteriales. • Vigilancia estricta de signos vitales • Control de convulsiones. .
 si la intoxicacion fue por via dermica ya. que el medio alcalino hidroliza el toxico. • No inducir vomito por el riesgo de broncoaspiracion. Ademas las presentaciones liquidas de. los plaguicidas muy frecuentemente contienen. hidrocarburos tipo kerosene que aumenta. el riesgo de producir neumonitis quimica. durante la emesis. • Lavado gastrico con abundante suero fisiologico. o solucion salina, si la ingesta fue hace. menos de 1 hora y protegiendo la via aerea. en caso de que el paciente tenga disminucion. del estado de conciencia. Administrar. carbon activado 1g/kg de peso cada 8 horas. para adsorber o atrapar el plaguicida y evitar. su absorcion. • La administracion de catartico salino no es. recomendada ya que puede exacerbar la gastroenteritis. causada por los organofosforados. o carbamatos. Ademas el cuadro colinergico. a su vez se acompana de diarrea. • Control y manejo de equilibrio acido-basico. del paciente. Administrar bicarbonato de sodio. segun requerimientos observados en los. gases arteriales. • Vigilancia estricta de signos vitales. • Control de convulsiones. .")
101
Atropina Antagonista competitivo de acetilcolina (++ recept. muscarinicos) Ver: disminución de secreciones y aumento de fc 0.02 – 0.1 mg/ Kg IV dosis 1 dosis inicial, se repite cada min hasta fc 80 lpm vs infusión continua. Mantenimiento: cada 30 min por 4 horas y después cada 6 Específico • Atropina: Es una amina terciaria por lo que atraviesa la barrera hemato-encefalica. Es la droga base para el tratamiento y su mecanismo de accion es ser antagonista competitivo con la acetilcolina principalmente en los receptores muscarinicos. La atropina tiene poco efecto en los receptores nicotinicos por tanto no antagoniza el sindrome nicotinico. Inicialmente se debe buscar “atropinizar” el paciente; los signos recomendados para vigilar la “atropinizacion” son: disminucion de secreciones y aumento de frecuencia cardiaca. Es muy importante que el paciente reciba oxigenacion previamente para que la fibra cardiaca pueda responder al efecto de la atropina. La miosis puede persistir aun con el paciente bien atropinizado, asi que no es un buen parametro de control. Dosis inicial: Se inicia atropina ampollas de 1 mg IV no diluidas, en cantidad determinada por el medico tratante segun la severidad del cuadro clinico (2, 5, 10 etc) y se continua con 1 mg IV cada 5 a 10 minutos, hasta alcanzar atropinizacion del paciente (disminucion de secreciones y aumento de frecuencia cardiaca por encima de 80 l/m). Dosis de mantenimiento: Una vez alcanzada la atropinizacion, se continua con 1 mg. IV cada media hora durante 3-4 horas pasandose posteriormente, segun respuesta del paciente a 1 mg cada 6 horas; la atropina debe mantenerse por el tiempo que lo requiera el paciente y hasta que cedan totalmente los sintomas. Dosis Pediátrica: 0.02 – 0.1 mg/ Kg IV dosis
 y se continua con 1 mg IV cada 5 a 10 minutos, hasta alcanzar atropinizacion del paciente (disminucion. de secreciones y aumento de frecuencia. cardiaca por encima de 80 l/m). Dosis de mantenimiento: Una vez alcanzada la atropinizacion, se continua. con 1 mg. IV cada media hora durante 3-4 horas. pasandose posteriormente, segun respuesta del. paciente a 1 mg cada 6 horas; la atropina debe. mantenerse por el tiempo que lo requiera el. paciente y hasta que cedan totalmente los sintomas. Dosis Pediátrica: 0.02 – 0.1 mg/ Kg IV dosis.")
102
Pralidoxima Reactiva acetilcolinesterasa fosforilada
Principalmente r. nicotínicos Mejora la contractilidad muscular en 10 a 40 min. Dosis: inicial 25 a 50 mg/kg (max 1 gr) iv en 30 min, repetir a em 1-2 horas si sigue com fasiculaciones Mantenimiento: 5-10mg/kg/hora. + Pralidoxima (2-PAM) u Oximas (Obidoxima). Es utilizada junto con la atropina en el manejo de pacientes con intoxicacion severa por organofosforados y en algunas oportunidades en casos severos por carbamatos, excepto en aldicarb, carbaryl y methomyl en los que su uso no tiene ningun beneficio. Su accion es reactivar la enzima acetilcolinesterasa fosforilada principalmente a nivel de receptores nicotinicos, mejorando la contractilidad muscular, dentro de los primeros 10 a 40 minutos despues de su administracion; presenta ademas sinergismo con la atropina en los receptores muscarinicos, por lo que siempre deben administrarse de manera conjunta. Su eficacia esta en relacion directa con la precocidad de su administracion (primeras 24 horas), pues el “envejecimiento” de la fosforilizacion aumenta la estabilidad del complejo enzima-fosforado. Durante su administracion el paciente preferiblemente debe estar bajo monitorizacion en una Unidad de Cuidados Intensivos. ADULTOS: Dosis inicial: Pralidoxima 1 gr. en 100 ml de solucion salina administrar via IV en 15-30 minutos. Dosis de Mantenimiento: Infusion de pralidoxima al 1 % (1gr de pralidoxima en 100 ml de SSN) pasar en infusion a mg/hora. NIÑOS: Dosis inicial: 25 a 50 mg/kg (maximo 1 gramo) intravenoso, administrados en 30 minutos. Se puede repetir la misma dosis en 1 a 2 horas, si el paciente persiste con fasciculaciones. Dosis de mantenimiento: Infusion de pralidoxima a 5-10mg/kg/hora. Durante el embarazo y la lactancia se considera un farmaco clase C. Con dosis terapeuticas en humanos, los efectos adversos son minimos. Con niveles mayores de 400 mg/ml se ha documentado vision borrosa, elevacion de la presion diastolica, diplopia, cefalea, nauseas estupor, hiperventilacion, hipotension, taquicardia, dolor muscular, mioclonias y agitacion. La administracion rapida intravenosa (>200 mg/minuto) puede causar paro cardiaco y respiratorio.
 iv en 30 min, repetir a em 1-2 horas si sigue com fasiculaciones. Mantenimiento: 5-10mg/kg/hora. + Pralidoxima (2-PAM) u Oximas. (Obidoxima). Es utilizada junto con la atropina en el manejo. de pacientes con intoxicacion severa por organofosforados. y en algunas oportunidades en. casos severos por carbamatos, excepto en aldicarb, carbaryl y methomyl en los que su uso no. tiene ningun beneficio. Su accion es reactivar. la enzima acetilcolinesterasa fosforilada principalmente. a nivel de receptores nicotinicos, mejorando la contractilidad muscular, dentro. de los primeros 10 a 40 minutos despues de su. administracion; presenta ademas sinergismo. con la atropina en los receptores muscarinicos, por lo que siempre deben administrarse de. manera conjunta. Su eficacia esta en relacion. directa con la precocidad de su administracion. (primeras 24 horas), pues el envejecimiento de la fosforilizacion aumenta la estabilidad del. complejo enzima-fosforado. Durante su administracion. el paciente preferiblemente debe. estar bajo monitorizacion en una Unidad de. Cuidados Intensivos. ADULTOS: Dosis inicial: Pralidoxima 1 gr. en 100 ml de. solucion salina administrar via IV en minutos. Dosis de Mantenimiento: Infusion de pralidoxima. al 1 % (1gr de pralidoxima en 100 ml. de SSN) pasar en infusion a mg/hora. NIÑOS: Dosis inicial: 25 a 50 mg/kg (maximo 1 gramo) intravenoso, administrados en 30 minutos. Se. puede repetir la misma dosis en 1 a 2 horas, si. el paciente persiste con fasciculaciones. Dosis de mantenimiento: Infusion de pralidoxima. a 5-10mg/kg/hora. Durante el embarazo y la lactancia se considera. un farmaco clase C. Con dosis terapeuticas en humanos, los efectos adversos. son minimos. Con niveles mayores de 400. mg/ml se ha documentado vision borrosa, elevacion. de la presion diastolica, diplopia, cefalea, nauseas. estupor, hiperventilacion, hipotension, taquicardia, dolor muscular, mioclonias y agitacion. La. administracion rapida intravenosa (>200 mg/minuto) puede causar paro cardiaco y respiratorio.")
103
Difenhidramina Coadyuvante para manejo de fasciculaciones 1 mg/k/12h
Antihistamínico, anti colinérgico, antimuscarinico • Difenhidramina (Benadryl): Se utiliza como coadyuvante en el tratamiento de intoxicacion por organofosforados y carbamatos. Su uso es de utilidad en el tratamiento de las fasciculaciones musculares, sobre las que no actua la atropina, por ser de efecto nicotinico. Se administra concomitantemente con atropina. Dosis: 50 mg (o 1 mg/kg de peso via oral) en jarabe por SNG con buenos resultados cada 8 horas en adultos y cada 12 horas en ninos.
: Se utiliza como coadyuvante en el tratamiento de. intoxicacion por organofosforados y carbamatos. Su uso es de utilidad en el tratamiento de las fasciculaciones. musculares, sobre las que no actua. la atropina, por ser de efecto nicotinico. Se administra. concomitantemente con atropina. Dosis: 50. mg (o 1 mg/kg de peso via oral) en jarabe por. SNG con buenos resultados cada 8 horas en adultos. y cada 12 horas en ninos.")
104
Metanol METHANOL Perspective
Methanol is a colorless, volatile, slightly sweet-tasting alcohol. It is a product of natural fermentation and originally was manufactured from the distillation of wood. Methanol currently is produced almost exclusively by synthetic pathways. Certain products found in the home may contain high concentrations of methanol, including antifreeze; windshield washer fluid; carburetor fluid; duplicator fluid; hobby engine fuel; gasohol; dry gas; Sterno; glass cleaners; and thinners for shellacs, lacquers, adhesives, and inks. Methanol is a precursor in the manufacture of plastics, films, and dyes. Methanol also is found in formalin and embalming fluid. Illicit alcohol production remains a global source of methanol poisoning from products such as chang'aa (Kenya), raki (Turkey), and tuica (Romania). Perspective METHANOL Although epidemic poisonings from methanol are reported occasionally, most exposures are sporadic. In 2002, of the 2822 cases of methanol poisoning reported to American poison centers, 86% were unintentional, 9% had moderate to major complications, and 18 resulted in fatalities. Treatment delay is associated with increased morbidity, making early recognition of clinical and laboratory clues crucial. Methanol is absorbed rapidly from the gastrointestinal tract, and blood levels peak 30 to 60 minutes after ingestion.[1] Transdermal and respiratory tract absorption also has resulted in toxicity, especially in infants. Certain occupations are at high risk for inhalational exposure to methanol, including painting, glazing, varnishing, lithography, and printing. At low serum concentrations, the elimination of methanol follows first-order kinetics; at concentrations after overdose, zero-order kinetics predominate. A prolonged half-life of 24 to 30 hours results, which may be extended even further by the concurrent ingestion of ethanol. First-order elimination prevails at high levels (>300 mg/dL), possibly as a result of enhanced pulmonary elimination. Small amounts of ingested methanol may be exceptionally toxic. In adults, the smallest lethal dose reported is 15 mL of 40% methanol; 4 mL of pure methanol has led to blindness. With appropriate, timely treatment, however, survival without loss of eyesight has been reported despite extremely high levels. From a pediatric perspective, the ingestion of only 1.5 mL of 100% methanol in a toddler (0.15 mL/kg) is sufficient to produce a toxic blood level of 20 mg/dL. Any suspected pediatric methanol ingestion warrants aggressive evaluation and treatment. Pharmacology and Metabolism Principles of Disease Methanol itself has little toxicity, producing less central nervous system (CNS) depression and inebriation than ethanol. Metabolites of the parent alcohol are extremely toxic, however. Although small amounts of methanol are eliminated via renal and pulmonary routes, 90% is metabolized hepatically. Methanol is oxidized by alcohol dehydrogenase (ADH) to formaldehyde, which is rapidly converted by aldehyde dehydrogenase to formic acid ( Figure ). Formic acid is the primary toxicant and accounts for much of the anion gap metabolic acidosis and ocular toxicity peculiar to methanol ingestion.[2] Through a folate-dependent pathway, formic acid is degraded to carbon dioxide and water. Figure 153-1 Metabolism of methanol. Optic neuropathy and putaminal necrosis are the two main complications of severe methanol poisoning. Long-term morbidity takes the form of visual impairment, including blindness, and parkinsonian motor dysfunction, characterized by hypokinesis and rigidity. Formic acid has a high affinity for iron and, as such, inhibits mitochondrial cytochrome oxidase, halting cellular respiration.[3] Methanol metabolism, in the cytosol and mitochondria, may account for a second mechanism of adenosine triphosphate depletion.[4] Lactate accumulation resulting from hypotension or seizures further compounds the metabolic acidosis predominantly caused by formate. Other mechanisms of toxicity involve increased lipid peroxidation, free radical formation, and impaired protective antioxidant reactions.[3] Severe dysfunction of subcellular metabolism from methanol also has been linked to significant disturbance in proteolytic-antiproteolytic balance.[5] Pathophysiology The primary sites of ocular injury are the retrolaminar optic nerve and retina. Selective myelin damage to the retrolaminar optic nerve in patients dying as a result of methanol toxicity has been seen at autopsy. Müller cells, the principal glial cells of retinal neurons and photoreceptors, have been proposed as the initial target in methanol-induced visual toxicity. It seems that they alone harbor the enzymes necessary to metabolize methanol to formate. Histopathologic correlates suggest that retinal cells develop intra-axonal swelling, calcium influx, mitochondrial destruction, and microtubular disruption. Ultimately, interference with transport of essential proteins from the retinal neuron cell body to the nerve fiber axoplasm results. Oligodendrocyte involvement results in myelin degeneration and leads to visual decrements. Acidosis may accelerate this process, by enhancing nonionic diffusion of formic acid into neurons and further increasing lactate production.[3] This self-perpetuating cycle of acidosis, termed circulus hypoxicus,[6] underscores the need for aggressive correction of pH to accomplish ion trapping of formate outside the CNS. Methanol adversely affects other areas of the CNS, specifically the basal ganglia. Bilateral, symmetric putaminal hypodensities, hemorrhages, or cystic lesions are characteristic, occurring in 13.5% of patients.[7] Necrosis is described in the subcortical white matter, spinal cord anterior horn cells, and cerebellum.[8] Acute signs and symptoms may be lacking or may take several days to develop, despite the presence of these radiographic findings. The cellular mechanisms of injury may be similar to the mechanisms of the ophthalmologic injury, but the reason for localization of neurologic damage to the basal ganglia is unknown.[7] Although some quantitative neuropathologic studies have shown high concentrations of formic acid within the putamen, others show that formate levels are not extraordinarily elevated in these areas compared with levels in the blood or other tissues. Massive edema adjacent to the putamen shown by magnetic resonance imaging (MRI) suggests a possible localized disruption of the blood-brain barrier. Other proposed mechanisms for the vulnerability of this region include the unique pattern of arterial blood supply and venous drainage and greater metabolic activity. With individual cases of methanol poisoning, the history may be unobtainable or unreliable. The diagnosis should be considered in patients with altered mental status, visual complaints, or metabolic acidosis or in patients with occupations at high risk for exposure. Because methanol is a poor substrate for ADH, a latency period exists between the time of ingestion and onset of visual or metabolic disturbance. The typical 12- to 24-hour latency may be shorter when large amounts are consumed or longer when ethanol is coingested (range 40 minutes to 72 hours).[1] In patients who present early, formate accumulation may be ongoing, with risk for significant toxicity despite being asymptomatic. When symptoms manifest, they are primarily neurologic, gastrointestinal, or ocular in nature. Clinical Features Although methanol is less inebriating than ethanol, early symptoms of methanol poisoning are depressed mental status, confusion, and ataxia. Nonspecific complaints of weakness, dizziness, anorexia, headache, and nausea develop. In severe cases, coma and seizures may be seen. Although vomiting and abdominal pain commonly result from mucosal irritation, the absence of gastrointestinal complaints does not rule out a serious ingestion.[1] Abdominal tenderness may be so severe that it suggests an acute surgical abdomen.[1] This may result from pancreatitis, and elevation of serum amylase is relatively common. Other authors have noted increased salivary amylase isoenzyme without pancreatic inflammation. Visual disturbances are seen in 50% of patients, and their development may precede or parallel that of other clinical symptoms.[6] Patients may complain of cloudy, blurred, indistinct, or misty vision or may note yellow spots or, rarely, photophobia. The most common acute field defect is a dense central scotoma.[9] Some patients compare their visual symptoms with ‘stepping out into a snowstorm,’ a complaint unique to methanol ingestion. Patients can have a complete lack of light perception and total loss of vision. On examination, optic disk hyperemia is seen at 18 to 48 hours after ingestion. Peripapillary retinal edema follows, is most striking in the nerve fiber layer along the vascular arcades, and only rarely involves the macula. [4] [9] Sluggishly reactive or fixed and dilated pupils indicate a poor prognosis. Pallor and cupping, indicative of optic atrophy, are late findings suggesting a poor prognosis for visual recovery. Occasionally, the fundus may appear normal, even in patients with visual symptoms. Compensatory tachypnea heralds the onset of metabolic acidosis, which often may be severe, with reported serum bicarbonate concentrations of less than 5 mEq/L and an arterial pH less than 7.0. Early tachycardia has been noted, but in general, cardiovascular abnormalities are rare.[1] Hypotension and bradycardia, when present, are preterminal findings.[10] Historically, death was described in association with a peculiar, abrupt cessation of respiration, rather than with cardiovascular collapse. [1] [11] Rarely, multiple organ failure develops.[10] Prognosis after methanol ingestion seems to correlate with the degree of acidosis, time to presentation, and initiation of treatment within 8 hours of exposure.[1] Poor prognosis is associated with coma, seizures, or arterial pH less than 7.0. [10] [12] Patients surviving the acute phase of toxicity may be left with permanent blindness or neurologic deficits, such as parkinsonism, toxic encephalopathy, polyneuropathy, cognitive dysfunction, transverse myelitis, primitive reflexes, or seizures. A severe anion gap metabolic acidosis is the hallmark of methanol ingestion. In some cases, this anion gap metabolic acidosis may be the only diagnostic clue. Because the onset of acidosis may be delayed 12 to 24 hours, the presence of a normal anion gap does not rule out methanol exposure. Absence of high anion gap acidosis has been described in cases with concomitant ethanol, lithium, or bromide ingestion. In methanol toxicity, this anion gap is due primarily to the presence of formic acid, with a variable contribution from lactic acid. Another classic laboratory finding in methanol toxicity is an elevated osmol gap. The osmolal gap is defined as follows: Diagnostic Strategies Osmol gap = measured serum osmolality - calculated serum osmolality The ‘normal’ osmol gap is often cited to be less than 10 mOsm/kg when the preceding equation is used. This is an arbitrary number, and there is considerable variability in baseline osmolal gaps in patients, particularly children.[13] If the osmol gap is significantly greater than 10 mOsm/kg, it may be a useful aid in the diagnosis of toxic alcohol ingestion. Caution should be taken, however, in ruling out toxic alcohol ingestion with a ‘normal’ osmol gap for several reasons. First, calculated serum osmolality results may vary among laboratories and must be done by the freezing point depression method. Also, delayed presentation after toxic alcohol ingestion may be associated with prior metabolism of most of the parent alcohol. Because only the parent compound is osmotically active, and because the charged metabolites are electrically balanced by sodium, there may be little or no osmol gap elevation in this setting. Finally, a toxic level of either methanol or ethylene glycol may be present with a gap of only 10 mOsm/kg. If there is clinical suspicion of toxic alcohol ingestion, direct measurement of the serum toxic alcohol level is necessary, and if not readily available, empiric treatment is warranted.[14] In addition to methanol, ethylene glycol, and isopropanol, other low-molecular-weight solutes may cause elevated osmol gaps, such as ethanol, acetone, propylene glycol, mannitol, glycerol, and ethyl ether. In addition to severe acidosis, rhabdomyolysis, pancreatitis, and metabolic derangements, such as hypomagnesemia, hypokalemia, and hypophosphatemia, are described. Calculated serum osmolality (mOsm/kg) = 2(Na+) + [BUN/2.8] + [glucose/18] + [ethanol (mg/dL)/4.6] Serum osmolality depends on the presence of low-molecular-weight solutes, primarily sodium, chloride, glucose, and blood urea nitrogen (BUN). One formula for calculating osmolality attributable to these solutes is as follows: Computed tomography may be indicated in an intoxicated patient with altered mental status. The characteristic finding of bilateral putaminal lesions suggests methanol poisoning, but this finding also may be seen with Leigh's syndrome, Wilson's disease, hypoxic-ischemic insult, encephalitis, and certain metabolic disorders. Ischemic necrosis, cerebral edema, or brain hemorrhages also may be noted. Follow-up scans may have prognostic value because parkinsonian features are unlikely to develop in patients whose putaminal lesions resolve within a short time frame.[15] MRI may detect optic neuropathy or putaminal aberrations from methanol intoxication. Methanol and ethylene glycol cause inebriation and are ingested as ethanol substitutes. The differential diagnosis of a patient with altered mental status includes hypoglycemia, head trauma, postictal state, carbon dioxide narcosis, hypoxia, infection, hepatic encephalopathy, other metabolic disorders, thiamine deficiency, endocrinopathy, drug abuse, and other poisoning. Patients who present with severe abdominal pain and altered mental status could slant the differential diagnosis toward a long list of intra-abdominal entities. When an anion gap acidosis is identified, however, the differential diagnosis must be tapered toward entities that cause this, and a primary decision must be made regarding whether the acidosis is a result of an ingested toxin or some other cause (e.g., mesenteric ischemia, diabetic ketoacidosis). Usually, the presence of an ingestion is admitted by the patient or strongly suspected by the providers, but this is not always the case, especially when the patient has depressed mental status of unknown cause. Causes of an elevated anion gap in patients without evidence of renal failure, hypotension, hypoxemia, diabetes, seizures, or alcoholism include methanol, ethylene glycol, paraldehyde, isoniazid, iron, salicylates, toluene, or lactic acidosis from myriad toxicants, including metformin, carbon monoxide, cyanide, and cocaine. Ethylene glycol and methanol may cause a ‘double gap’ (i.e., an osmol gap in addition to the anion gap). Other substances that contribute to an elevated osmol gap include isopropyl alcohol, ethanol, propylene glycol, mannitol, glycerol, and ethyl ether. Other situations in which double-gap acidosis may be encountered include diabetic ketoacidosis; alcoholic ketoacidosis; acetonitrile, methanol, ethylene glycol, and propylene glycol toxicity; multiple organ failure; chronic renal failure; and critical illness.[16] Hyperlipidemia and hyperproteinemia, by decreasing the measured sodium concentration, can increase the osmolal gap. Characteristically, isopropanol does not cause an increased anion gap. Differential Considerations Certain unusual characteristics of methanol and ethylene glycol intoxication lead to the specific diagnosis. The presence of ocular complaints unique to methanol poisoning is a valuable clue. Ethylene glycol ingestion often is associated with calcium oxalate crystalluria, which is not seen in methanol ingestion. Ultimately, the definitive diagnosis depends on the identification of the parent alcohol in the blood by laboratory tests that may not be routinely available. It is often necessary to start treatment based on clinical suspicion alone. Because the initial treatment for methanol and ethylene glycol is almost identical, identification of the specific toxic alcohol is not crucial to the initiation of therapy. See the section on ethylene glycol management. Management and Disposition Metanol
, raki (Turkey), and tuica (Romania). Perspective. METHANOL. Although epidemic poisonings from methanol are reported occasionally, most exposures are sporadic. In 2002, of the 2822 cases of methanol poisoning reported to American poison centers, 86% were unintentional, 9% had moderate to major complications, and 18 resulted in fatalities. Treatment delay is associated with increased morbidity, making early recognition of clinical and laboratory clues crucial. Methanol is absorbed rapidly from the gastrointestinal tract, and blood levels peak 30 to 60 minutes after ingestion.[1] Transdermal and respiratory tract absorption also has resulted in toxicity, especially in infants. Certain occupations are at high risk for inhalational exposure to methanol, including painting, glazing, varnishing, lithography, and printing. At low serum concentrations, the elimination of methanol follows first-order kinetics; at concentrations after overdose, zero-order kinetics predominate. A prolonged half-life of 24 to 30 hours results, which may be extended even further by the concurrent ingestion of ethanol. First-order elimination prevails at high levels (>300 mg/dL), possibly as a result of enhanced pulmonary elimination. Small amounts of ingested methanol may be exceptionally toxic. In adults, the smallest lethal dose reported is 15 mL of 40% methanol; 4 mL of pure methanol has led to blindness. With appropriate, timely treatment, however, survival without loss of eyesight has been reported despite extremely high levels. From a pediatric perspective, the ingestion of only 1.5 mL of 100% methanol in a toddler (0.15 mL/kg) is sufficient to produce a toxic blood level of 20 mg/dL. Any suspected pediatric methanol ingestion warrants aggressive evaluation and treatment. Pharmacology and Metabolism. Principles of Disease. Methanol itself has little toxicity, producing less central nervous system (CNS) depression and inebriation than ethanol. Metabolites of the parent alcohol are extremely toxic, however. Although small amounts of methanol are eliminated via renal and pulmonary routes, 90% is metabolized hepatically. Methanol is oxidized by alcohol dehydrogenase (ADH) to formaldehyde, which is rapidly converted by aldehyde dehydrogenase to formic acid ( Figure ). Formic acid is the primary toxicant and accounts for much of the anion gap metabolic acidosis and ocular toxicity peculiar to methanol ingestion.[2] Through a folate-dependent pathway, formic acid is degraded to carbon dioxide and water. Figure Metabolism of methanol. Optic neuropathy and putaminal necrosis are the two main complications of severe methanol poisoning. Long-term morbidity takes the form of visual impairment, including blindness, and parkinsonian motor dysfunction, characterized by hypokinesis and rigidity. Formic acid has a high affinity for iron and, as such, inhibits mitochondrial cytochrome oxidase, halting cellular respiration.[3] Methanol metabolism, in the cytosol and mitochondria, may account for a second mechanism of adenosine triphosphate depletion.[4] Lactate accumulation resulting from hypotension or seizures further compounds the metabolic acidosis predominantly caused by formate. Other mechanisms of toxicity involve increased lipid peroxidation, free radical formation, and impaired protective antioxidant reactions.[3] Severe dysfunction of subcellular metabolism from methanol also has been linked to significant disturbance in proteolytic-antiproteolytic balance.[5] Pathophysiology. The primary sites of ocular injury are the retrolaminar optic nerve and retina. Selective myelin damage to the retrolaminar optic nerve in patients dying as a result of methanol toxicity has been seen at autopsy. Müller cells, the principal glial cells of retinal neurons and photoreceptors, have been proposed as the initial target in methanol-induced visual toxicity. It seems that they alone harbor the enzymes necessary to metabolize methanol to formate. Histopathologic correlates suggest that retinal cells develop intra-axonal swelling, calcium influx, mitochondrial destruction, and microtubular disruption. Ultimately, interference with transport of essential proteins from the retinal neuron cell body to the nerve fiber axoplasm results. Oligodendrocyte involvement results in myelin degeneration and leads to visual decrements. Acidosis may accelerate this process, by enhancing nonionic diffusion of formic acid into neurons and further increasing lactate production.[3] This self-perpetuating cycle of acidosis, termed circulus hypoxicus,[6] underscores the need for aggressive correction of pH to accomplish ion trapping of formate outside the CNS. Methanol adversely affects other areas of the CNS, specifically the basal ganglia. Bilateral, symmetric putaminal hypodensities, hemorrhages, or cystic lesions are characteristic, occurring in 13.5% of patients.[7] Necrosis is described in the subcortical white matter, spinal cord anterior horn cells, and cerebellum.[8] Acute signs and symptoms may be lacking or may take several days to develop, despite the presence of these radiographic findings. The cellular mechanisms of injury may be similar to the mechanisms of the ophthalmologic injury, but the reason for localization of neurologic damage to the basal ganglia is unknown.[7] Although some quantitative neuropathologic studies have shown high concentrations of formic acid within the putamen, others show that formate levels are not extraordinarily elevated in these areas compared with levels in the blood or other tissues. Massive edema adjacent to the putamen shown by magnetic resonance imaging (MRI) suggests a possible localized disruption of the blood-brain barrier. Other proposed mechanisms for the vulnerability of this region include the unique pattern of arterial blood supply and venous drainage and greater metabolic activity. With individual cases of methanol poisoning, the history may be unobtainable or unreliable. The diagnosis should be considered in patients with altered mental status, visual complaints, or metabolic acidosis or in patients with occupations at high risk for exposure. Because methanol is a poor substrate for ADH, a latency period exists between the time of ingestion and onset of visual or metabolic disturbance. The typical 12- to 24-hour latency may be shorter when large amounts are consumed or longer when ethanol is coingested (range 40 minutes to 72 hours).[1] In patients who present early, formate accumulation may be ongoing, with risk for significant toxicity despite being asymptomatic. When symptoms manifest, they are primarily neurologic, gastrointestinal, or ocular in nature. Clinical Features. Although methanol is less inebriating than ethanol, early symptoms of methanol poisoning are depressed mental status, confusion, and ataxia. Nonspecific complaints of weakness, dizziness, anorexia, headache, and nausea develop. In severe cases, coma and seizures may be seen. Although vomiting and abdominal pain commonly result from mucosal irritation, the absence of gastrointestinal complaints does not rule out a serious ingestion.[1] Abdominal tenderness may be so severe that it suggests an acute surgical abdomen.[1] This may result from pancreatitis, and elevation of serum amylase is relatively common. Other authors have noted increased salivary amylase isoenzyme without pancreatic inflammation. Visual disturbances are seen in 50% of patients, and their development may precede or parallel that of other clinical symptoms.[6] Patients may complain of cloudy, blurred, indistinct, or misty vision or may note yellow spots or, rarely, photophobia. The most common acute field defect is a dense central scotoma.[9] Some patients compare their visual symptoms with ‘stepping out into a snowstorm,’ a complaint unique to methanol ingestion. Patients can have a complete lack of light perception and total loss of vision. On examination, optic disk hyperemia is seen at 18 to 48 hours after ingestion. Peripapillary retinal edema follows, is most striking in the nerve fiber layer along the vascular arcades, and only rarely involves the macula. [4] [9] Sluggishly reactive or fixed and dilated pupils indicate a poor prognosis. Pallor and cupping, indicative of optic atrophy, are late findings suggesting a poor prognosis for visual recovery. Occasionally, the fundus may appear normal, even in patients with visual symptoms. Compensatory tachypnea heralds the onset of metabolic acidosis, which often may be severe, with reported serum bicarbonate concentrations of less than 5 mEq/L and an arterial pH less than 7.0. Early tachycardia has been noted, but in general, cardiovascular abnormalities are rare.[1] Hypotension and bradycardia, when present, are preterminal findings.[10] Historically, death was described in association with a peculiar, abrupt cessation of respiration, rather than with cardiovascular collapse. [1] [11] Rarely, multiple organ failure develops.[10] Prognosis after methanol ingestion seems to correlate with the degree of acidosis, time to presentation, and initiation of treatment within 8 hours of exposure.[1] Poor prognosis is associated with coma, seizures, or arterial pH less than 7.0. [10] [12] Patients surviving the acute phase of toxicity may be left with permanent blindness or neurologic deficits, such as parkinsonism, toxic encephalopathy, polyneuropathy, cognitive dysfunction, transverse myelitis, primitive reflexes, or seizures. A severe anion gap metabolic acidosis is the hallmark of methanol ingestion. In some cases, this anion gap metabolic acidosis may be the only diagnostic clue. Because the onset of acidosis may be delayed 12 to 24 hours, the presence of a normal anion gap does not rule out methanol exposure. Absence of high anion gap acidosis has been described in cases with concomitant ethanol, lithium, or bromide ingestion. In methanol toxicity, this anion gap is due primarily to the presence of formic acid, with a variable contribution from lactic acid. Another classic laboratory finding in methanol toxicity is an elevated osmol gap. The osmolal gap is defined as follows: Diagnostic Strategies. Osmol gap = measured serum osmolality - calculated serum osmolality. The ‘normal’ osmol gap is often cited to be less than 10 mOsm/kg when the preceding equation is used. This is an arbitrary number, and there is considerable variability in baseline osmolal gaps in patients, particularly children.[13] If the osmol gap is significantly greater than 10 mOsm/kg, it may be a useful aid in the diagnosis of toxic alcohol ingestion. Caution should be taken, however, in ruling out toxic alcohol ingestion with a ‘normal’ osmol gap for several reasons. First, calculated serum osmolality results may vary among laboratories and must be done by the freezing point depression method. Also, delayed presentation after toxic alcohol ingestion may be associated with prior metabolism of most of the parent alcohol. Because only the parent compound is osmotically active, and because the charged metabolites are electrically balanced by sodium, there may be little or no osmol gap elevation in this setting. Finally, a toxic level of either methanol or ethylene glycol may be present with a gap of only 10 mOsm/kg. If there is clinical suspicion of toxic alcohol ingestion, direct measurement of the serum toxic alcohol level is necessary, and if not readily available, empiric treatment is warranted.[14] In addition to methanol, ethylene glycol, and isopropanol, other low-molecular-weight solutes may cause elevated osmol gaps, such as ethanol, acetone, propylene glycol, mannitol, glycerol, and ethyl ether. In addition to severe acidosis, rhabdomyolysis, pancreatitis, and metabolic derangements, such as hypomagnesemia, hypokalemia, and hypophosphatemia, are described. Calculated serum osmolality (mOsm/kg) = 2(Na+) + [BUN/2.8] + [glucose/18] + [ethanol (mg/dL)/4.6] Serum osmolality depends on the presence of low-molecular-weight solutes, primarily sodium, chloride, glucose, and blood urea nitrogen (BUN). One formula for calculating osmolality attributable to these solutes is as follows: Computed tomography may be indicated in an intoxicated patient with altered mental status. The characteristic finding of bilateral putaminal lesions suggests methanol poisoning, but this finding also may be seen with Leigh s syndrome, Wilson s disease, hypoxic-ischemic insult, encephalitis, and certain metabolic disorders. Ischemic necrosis, cerebral edema, or brain hemorrhages also may be noted. Follow-up scans may have prognostic value because parkinsonian features are unlikely to develop in patients whose putaminal lesions resolve within a short time frame.[15] MRI may detect optic neuropathy or putaminal aberrations from methanol intoxication. Methanol and ethylene glycol cause inebriation and are ingested as ethanol substitutes. The differential diagnosis of a patient with altered mental status includes hypoglycemia, head trauma, postictal state, carbon dioxide narcosis, hypoxia, infection, hepatic encephalopathy, other metabolic disorders, thiamine deficiency, endocrinopathy, drug abuse, and other poisoning. Patients who present with severe abdominal pain and altered mental status could slant the differential diagnosis toward a long list of intra-abdominal entities. When an anion gap acidosis is identified, however, the differential diagnosis must be tapered toward entities that cause this, and a primary decision must be made regarding whether the acidosis is a result of an ingested toxin or some other cause (e.g., mesenteric ischemia, diabetic ketoacidosis). Usually, the presence of an ingestion is admitted by the patient or strongly suspected by the providers, but this is not always the case, especially when the patient has depressed mental status of unknown cause. Causes of an elevated anion gap in patients without evidence of renal failure, hypotension, hypoxemia, diabetes, seizures, or alcoholism include methanol, ethylene glycol, paraldehyde, isoniazid, iron, salicylates, toluene, or lactic acidosis from myriad toxicants, including metformin, carbon monoxide, cyanide, and cocaine. Ethylene glycol and methanol may cause a ‘double gap’ (i.e., an osmol gap in addition to the anion gap). Other substances that contribute to an elevated osmol gap include isopropyl alcohol, ethanol, propylene glycol, mannitol, glycerol, and ethyl ether. Other situations in which double-gap acidosis may be encountered include diabetic ketoacidosis; alcoholic ketoacidosis; acetonitrile, methanol, ethylene glycol, and propylene glycol toxicity; multiple organ failure; chronic renal failure; and critical illness.[16] Hyperlipidemia and hyperproteinemia, by decreasing the measured sodium concentration, can increase the osmolal gap. Characteristically, isopropanol does not cause an increased anion gap. Differential Considerations. Certain unusual characteristics of methanol and ethylene glycol intoxication lead to the specific diagnosis. The presence of ocular complaints unique to methanol poisoning is a valuable clue. Ethylene glycol ingestion often is associated with calcium oxalate crystalluria, which is not seen in methanol ingestion. Ultimately, the definitive diagnosis depends on the identification of the parent alcohol in the blood by laboratory tests that may not be routinely available. It is often necessary to start treatment based on clinical suspicion alone. Because the initial treatment for methanol and ethylene glycol is almost identical, identification of the specific toxic alcohol is not crucial to the initiation of therapy. See the section on ethylene glycol management. Management and Disposition. Metanol.")
105
Alcohol industrial Alcohol de cocina Pinturas Solventes Adhesivos
Licores adulterados alcohol industrial, alcohol de cocina o alcohol de “reverbero”. pinturas, soluciones de limpieza, colorantes, resinas, adhesivos, productos de impermeabilización, cristales inastillables y productos fotográficos y otros materiales.

106
Absorción Piel: el que predomina en niños Mucosas Pulmones
“ En Bogotá, según el Instituto Nacional de Medicina Legal, durante los años se reportaron 99 muertes por intoxicación metílica comprobadas por análisis toxicológico; esto demuestra la gran magnitud del problema en nuestro medio. “ En el caso de los niños la vía de ingreso es la piel, pues las madres tienen la costumbre popular de hacer fricciones de alcohol como tratamiento de síntomas comunes, usando alcohol de cocina o industrial por desconocimiento o ignorancia. En Bogotá, según el Instituto Nacional de Medicina Legal, durante los años se reportaron 99 muertes por intoxicación metílica comprobadas por análisis toxicológico; esto demuestra la gran magnitud del problema en nuestro medio. inhalación de vapores o absorción por la piel por manipulación inadecuada. principalmente por adulteración de licores que se expenden comercialmente. El valor comercial con respecto al alcohol etílico es aproximadamente tres veces menor y el metanol se comercia libremente sin ningún tipo de restricción legal

107
Dosis tóxica Tóxica: 150 mm/kg Dosis letal: 30-240 cc 40 mg/dl letal
La dosis letal del metanol está estimada en mL ( gramos). La dosis tóxica mínima es aproximadamente de 100 mg/kg. Se pueden encontrar niveles elevados de metanol en sangre luego de exposición dérmica extensa o por inhalación. Una concentración sérica de 40 mg% es mortal.
. La dosis tóxica. mínima es aproximadamente de 100 mg/kg. Se pueden encontrar niveles elevados de metanol. en sangre luego de exposición dérmica. extensa o por inhalación. Una concentración. sérica de 40 mg% es mortal.")
108
Farmacocinética Absorción rápida
Preferencia por órganos con más agua: cerebro, humor acuoso y riñon. Metabolismo LENTO por alcohol deshidrogenasa (20 veces mas preferencia por el etanol) Eliminación renal y pulmonar perfunde rápidamente todos los órganos, especialmente aquellos ricos en agua como cerebro, humor acuoso y riñón. FARMACOCINÉTICA El metanol es absorbido y rápidamente distribuido por el agua del cuerpo. No se une a proteínas. Es metabolizado lentamente por la alcohol-deshidrogenasa. La vida media oscila entre 2 y 24 horas. Apenas cerca del 3% es excretado sin cambios por el riñón y menos del 10% a través del pulmón.
 Eliminación renal y pulmonar. perfunde rápidamente. todos los órganos, especialmente. aquellos ricos en agua como cerebro, humor. acuoso y riñón. FARMACOCINÉTICA. El metanol es absorbido y rápidamente distribuido. por el agua del cuerpo. No se une a. proteínas. Es metabolizado lentamente por la. alcohol-deshidrogenasa. La vida media oscila. entre 2 y 24 horas. Apenas cerca del 3% es. excretado sin cambios por el riñón y menos. del 10% a través del pulmón.")
110
Mecanismos de toxicidad
Metanol---formaldehido-----acido fórmico Acidosis metabólica (asociado a metab anaerobio) Ceguera: formaldehido La velocidad de metabolismo y eliminación de metanol es 7 a 10 veces más lenta que la de etanol por lo que permanece más tiempo en el organismo y hay un período de latencia característico entre 6 y 30 horas. La degradación de metanol en sus metabolitos tóxicos, especialmente el formato tiene lugar en la retina, en donde se cree que la energía lumínica (consumo de oxígeno) desempeña un papel fundamental. Como en el caso de alcohol etílico, existen grados clínico de intoxicación, que se relacionan con sus respectivas metanolemias así: Grado l: < de 10 mg%: Cefalea, náuseas, malestar general, ebriedad. En el caso de nuestro paciente, esta fase podría estar representada por la sintomatología presentada a su ingreso al servicio de urgencias. Grado ll: De 10 a 50 mg %: Comienza a observarse depresión del SNC, agitación psicomotora, signos de acidosis metabólica leve, pueden presentarse trastornos de la visión. Grado lll: >50 mg %: Se explica con el cuadro clínico del reingreso del paciente ya descrito. Es importante destacar que después de la sintomatología inicial hay un período libre de hasta 30 horas durante el cual se generan la acidosis y el daño ocular.
 Ceguera: formaldehido. La velocidad de metabolismo y eliminación de metanol es 7 a 10 veces más. lenta que la de etanol. por lo que permanece más tiempo en el organismo y. hay un período de latencia característico entre 6 y 30 horas. La degradación de. metanol en sus metabolitos tóxicos, especialmente el formato tiene lugar en la. retina, en donde se cree que la energía lumínica (consumo de oxígeno) desempeña un papel fundamental. Como en el caso de alcohol etílico, existen. grados clínico de intoxicación, que se relacionan con sus respectivas. metanolemias así: Grado l: < de 10 mg%: Cefalea, náuseas, malestar general, ebriedad. En el. caso de nuestro paciente, esta fase podría estar representada por la. sintomatología presentada a su ingreso al servicio de urgencias. Grado ll: De 10 a 50 mg %: Comienza a observarse depresión del SNC, agitación psicomotora, signos de acidosis metabólica leve, pueden presentarse. trastornos de la visión. Grado lll: >50 mg %: Se explica con el cuadro clínico del reingreso del paciente. ya descrito. Es importante destacar que después de la sintomatología inicial hay. un período libre de hasta 30 horas durante el cual se generan la acidosis y el daño. ocular.")
111
Mecanismos de toxicidad
La velocidad de metabolismo y eliminación de metanol es 7 a 10 veces más lenta que la de etanol por lo que permanece más tiempo en el organismo y hay un período de latencia característico entre 6 y 30 horas. La degradación de metanol en sus metabolitos tóxicos, especialmente el formato tiene lugar en la retina, en donde se cree que la energía lumínica (consumo de oxígeno) desempeña un papel fundamental. Como en el caso de alcohol etílico, existen grados clínico de intoxicación, que se relacionan con sus respectivas metanolemias así: Grado l: < de 10 mg%: Cefalea, náuseas, malestar general, ebriedad. En el caso de nuestro paciente, esta fase podría estar representada por la sintomatología presentada a su ingreso al servicio de urgencias. Grado ll: De 10 a 50 mg %: Comienza a observarse depresión del SNC, agitación psicomotora, signos de acidosis metabólica leve, pueden presentarse trastornos de la visión. Grado lll: >50 mg %: Se explica con el cuadro clínico del reingreso del paciente ya descrito. Es importante destacar que después de la sintomatología inicial hay un período libre de hasta 30 horas durante el cual se generan la acidosis y el daño ocular.
 desempeña un papel fundamental. Como en el caso de alcohol etílico, existen. grados clínico de intoxicación, que se relacionan con sus respectivas. metanolemias así: Grado l: < de 10 mg%: Cefalea, náuseas, malestar general, ebriedad. En el. caso de nuestro paciente, esta fase podría estar representada por la. sintomatología presentada a su ingreso al servicio de urgencias. Grado ll: De 10 a 50 mg %: Comienza a observarse depresión del SNC, agitación psicomotora, signos de acidosis metabólica leve, pueden presentarse. trastornos de la visión. Grado lll: >50 mg %: Se explica con el cuadro clínico del reingreso del paciente. ya descrito. Es importante destacar que después de la sintomatología inicial hay. un período libre de hasta 30 horas durante el cual se generan la acidosis y el daño. ocular.")
112
Grados de intoxicación
Grado l: < de 10 mg/dl: Cefalea, náuseas, malestar general, Grado ll: De 10 a 50 mg /dl: depresión del SNC, agitación acidosis metabólica leve, alt. de la visión. Grado lll: >50 mg /dl: después de latencia hasta 30 h. acidosis y el daño ocular.

113
Progresión clínica Primeras horas: embriaguez, cefalea, anion gap elevado 30 horas después: acidosis met. Severa, alt visuales, convulsiones, coma, ceguera. Fondo de ojo con hiperemia del disco óptico o papiledema. Midriasis precoz y no reactiva son signos de mal pronóstico.

114
Diagnóstico HC, EF, PARACLINICOS: gases, brecha aniónica,
Metanol en sangre y sus metabolitos: >20 mg/dL tóxicos >40 mg/dL fatales. Si la consulta es 30 horas después no se descarta la intox. por niveles bajos. Metanol o formaldehido en orina Electrolitos, glucosa, BUN, creatinina, osmolaridad serica, niveles de etanol, lactato

115
Tratamiento EVITAR EL METABOLISMO DEL METANOL
Vía aérea y ventilación, saturación Vendaje ocular Si convulsiones manejarlas Corrección de acidosis con HCO3 Na

116
Etanol terapia La idea es saturar la alcohol deshidrohenasa con etanol
Para: anion GAP: mayor de 5, Ac, met + anion GAP mayor , metanol en sangre positivo, formaldehido en sangre positivo Dosis de carga de cc de alcohol absoluto/kg. Mantenimiento: 0.16 cc/kg/h por 72h RIESGO DE HIPOGLICEMIA PARA DAR VO SE ASUME BIODISPONIBILIDAD DEL 100% (Se cuenta con ampollas de alcohol absoluto al 96% de 2mL, 5mL, 10 mL. Para aplicación 10% o menos para evitar que la mezcla cause en dextrosa al 5% a una concentración de intravenosa se debe diluir la cantidad necesaria flebitis por el efecto irritante del alcohol etílico ampollas, se puede suministrar por vía oral Ante la carencia de alcohol etílico puro o en concentrado). licor en sus diferentes presentaciones disponibles correspondientes. En ancianos y en niños estas aguardiente (30-35%) haciendo los cálculos como el whisky o vodka (40-45%), dosis de etanol pueden producir importante de glucosa y suministrar suero glucosado. hacer control periódico de los niveles sanguíneos disminución de la glicemia, por lo que se debe Ejemplo: paciente de 60 kilos de peso, requerirá en 600 mL de DAD al 5%, para pasar IV de carga, los cuales se deben diluir mínimo 60 mL de alcohol absoluto como dosis en 30 minutos; si no hay presentación en ampollas en cuenta que si 100 mL de aguardiente al 30% por sonda nasogástrica (SNG), teniendo se administrará 200 mL de aguardiente contienen 30 mL de alcohol absoluto, los 60 será de 0,16 mL por 60 kg cada hora, lo mL. La dosis de mantenimiento por vía intravenosa ml que necesitamos están contenidos en 200 o a 33,3 mL (una onza o copita) de que equivale a 9,6 mL en los líquidos de mantenimiento aguardiente por SNG cada hora.
. licor en sus diferentes presentaciones disponibles. correspondientes. En ancianos y en niños estas. aguardiente (30-35%) haciendo los cálculos. como el whisky o vodka (40-45%), dosis de etanol pueden producir importante. de glucosa y suministrar suero glucosado. hacer control periódico de los niveles sanguíneos. disminución de la glicemia, por lo que se debe. Ejemplo: paciente de 60 kilos de peso, requerirá. en 600 mL de DAD al 5%, para pasar IV. de carga, los cuales se deben diluir mínimo. 60 mL de alcohol absoluto como dosis. en 30 minutos; si no hay presentación en ampollas. en cuenta que si 100 mL de aguardiente. al 30% por sonda nasogástrica (SNG), teniendo. se administrará 200 mL de aguardiente. contienen 30 mL de alcohol absoluto, los 60. será de 0,16 mL por 60 kg cada hora, lo. mL. La dosis de mantenimiento por vía intravenosa. ml que necesitamos están contenidos en 200. o a 33,3 mL (una onza o copita) de. que equivale a 9,6 mL en los líquidos de mantenimiento. aguardiente por SNG cada hora.")
117
OTRAS MEDIDAS Lavado gástrico Diuresis forzada: furosemida
Balance estricto LA/LE Tiamina Ac folico 1 mg/kg iv /4h por 6 dosis: favorece eliminación de ac formico Tiamina: cofactor importante para el metabolismo de los carbohidratos, está indicada en intoxicación por alcohol etílico y en etiloterapia. Se administran 100 mg IV cada ocho horas durante tratamiento IV y se continúa con 300 mg/día vía oral por 10 días más. Ácido fólico: es parte del complejo vitamínico B. Puede favorecer el metabolismo del ácido fórmico y su eliminación. El ser humano no cuenta con suficiente ácido fólico para lograr ese paso metabólico, por lo cual se requieren dosis altas de 50 mg (niños 1mg/kg) intravenosos cada cuatro horas por seis dosis mínimo. El tratamiento en casos graves debe ser realizado interdisciplinariamente por el toxicólogo, el nefrólogo y el equipo de la unidad de cuidado intensivo.
 intravenosos cada cuatro horas por seis dosis. mínimo. El tratamiento en casos graves debe ser realizado. interdisciplinariamente por el toxicólogo, el nefrólogo y el equipo de la unidad de cuidado. intensivo.")
118
HEMODIÁLISIS Ph 7.2 o menor Anion GAP mayor 10 HCO3: MENOR 10
METANOL MAYOR DE 40MG/DL Compromiso visual importante Deterioro progresivo a pesar del manejo El esquema terapéutico aquí sugerido considera una recomendación Grado B. • Lavado gástrico exhaustivo, previa hidratación. • Mantener estricto equilibrio hidroelectrolítico y acidobásico. • Mantener diuresis con el empleo de un diurético osmótico tipo osmorín (debe mantenerse mientras la metanolemia sea positiva, con un estricto control de líquidos y electrolitos). • Gasimetría sanguínea acompañada de ionograma. • Prevención de la broncoaspiración y manejo de sus complicaciones en caso de presentarse. • Soporte ventilatorio si es necesario. • Corrección de acidosis metabólica. • Etiloterapia: Se emplea el alcohol etílico en concentraciones del 94 ó 98 % para uso IV, a una dosis de 130 mg por Kg, en infusión permanente para 12 horas (se aconseja control permanente de etanol y metanolemias). En caso de no disponerse de esta presentación puede administrase alcohol etílico (Whisky o Vodka) vía oral o por sonda nasogástrica, a una dosis de 0.5 cc por Kg de peso cada 4 horas, hasta negativizar la metanolemia (esta vía de administración se asocia frecuentemente con problemas de gastritis que deben ser previstos y/o manejados por el médico). • Vendaje ocular bilateral: Mientras exista metanol positivo debe realizarse, ya que busca neutralizar el papel de la energía lumínica en la biotransformación del metanol en ácido fórmico a nivel de la retina. • Acido fólico: 50 mg IV cada 4 horas (aumenta la eliminación de formatos). • Corticosteroides: Aunque su uso sigue siendo tema de controversia, en nuestra práctica hemos visto que resulta de utilidad, sobre todo en lo relacionado con la prevención y manejo del edema cerebral y ocular. • Hemodiálisis: En condiciones ideales, es el tratamiento de elección en todo caso de intoxicación comprobada por metanol. • 4-Metilpirazol (4-MP) inhibe el alcohol deshidrogenasa (disponibilidad limitada).
. • Gasimetría sanguínea acompañada de ionograma. • Prevención de la broncoaspiración y manejo de sus complicaciones en. caso de presentarse. • Soporte ventilatorio si es necesario. • Corrección de acidosis metabólica. • Etiloterapia: Se emplea el alcohol etílico en concentraciones del 94 ó. 98 % para uso IV, a una dosis de 130 mg por Kg, en infusión permanente. para 12 horas (se aconseja control permanente de etanol y metanolemias). En caso de no disponerse de esta presentación puede. administrase alcohol etílico (Whisky o Vodka) vía oral o por sonda. nasogástrica, a una dosis de 0.5 cc por Kg de peso cada 4 horas, hasta. negativizar la metanolemia (esta vía de administración se asocia. frecuentemente con problemas de gastritis que deben ser previstos y/o. manejados por el médico). • Vendaje ocular bilateral: Mientras exista metanol positivo debe realizarse, ya que busca neutralizar el papel de la energía lumínica en la. biotransformación del metanol en ácido fórmico a nivel de la retina. • Acido fólico: 50 mg IV cada 4 horas (aumenta la eliminación de. formatos). • Corticosteroides: Aunque su uso sigue siendo tema de controversia, en. nuestra práctica hemos visto que resulta de utilidad, sobre todo en lo. relacionado con la prevención y manejo del edema cerebral y ocular. • Hemodiálisis: En condiciones ideales, es el tratamiento de elección en. todo caso de intoxicación comprobada por metanol. • 4-Metilpirazol (4-MP) inhibe el alcohol deshidrogenasa (disponibilidad. limitada).")
119
G R A C I A S Gracias!

Presentaciones similares
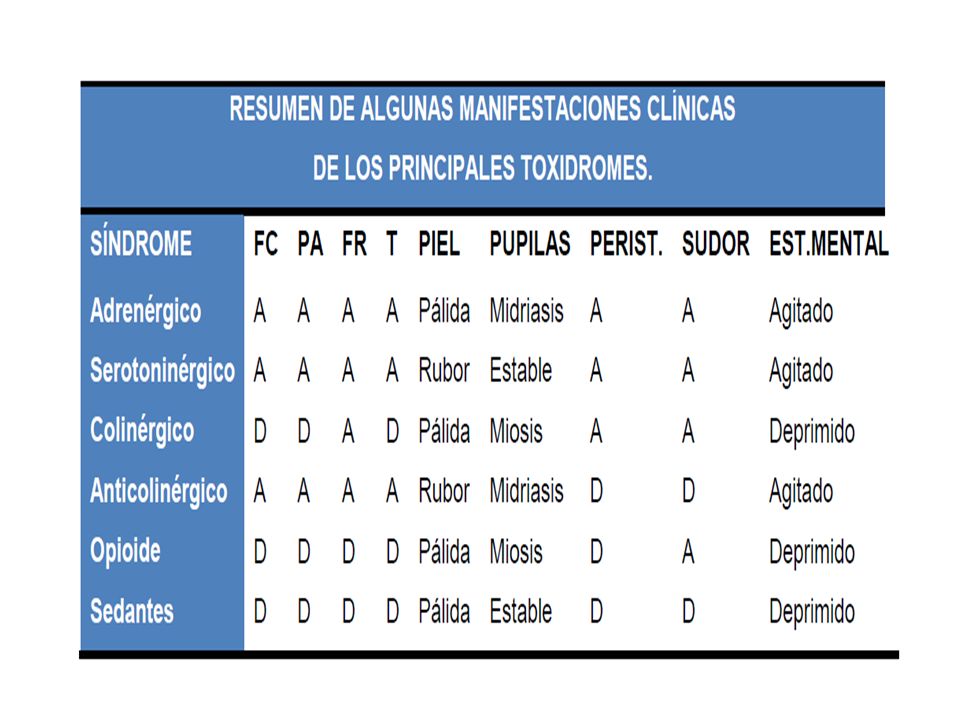

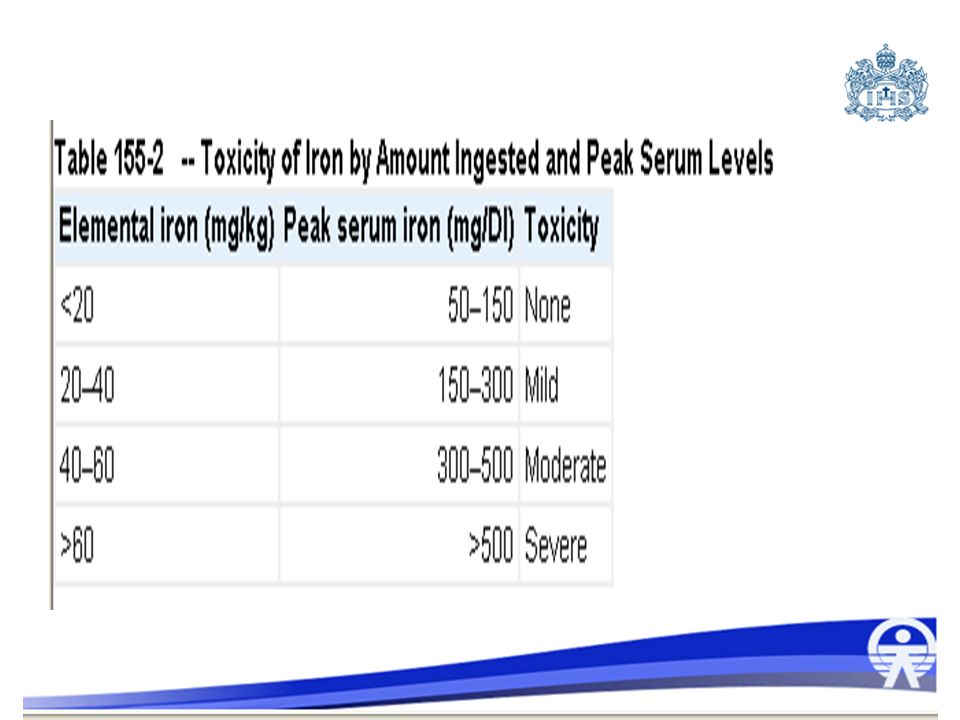
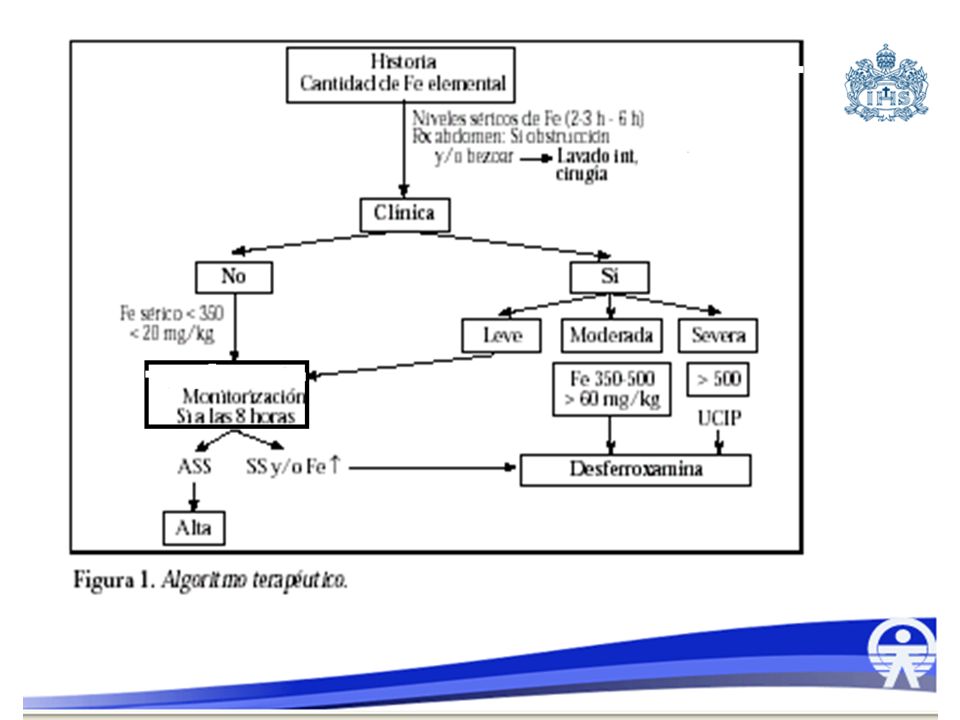
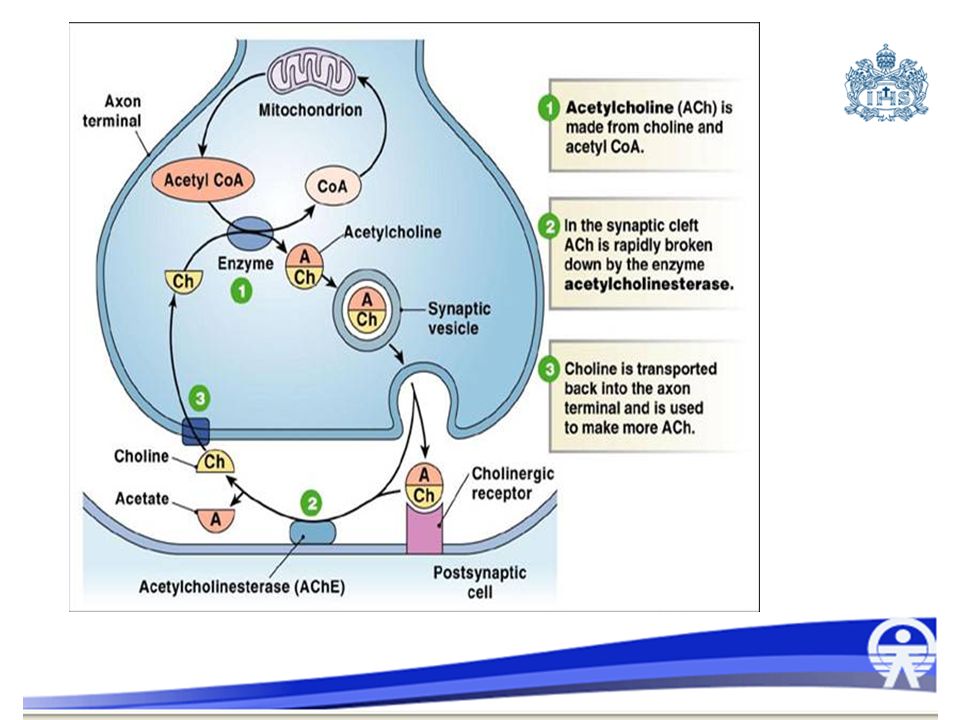
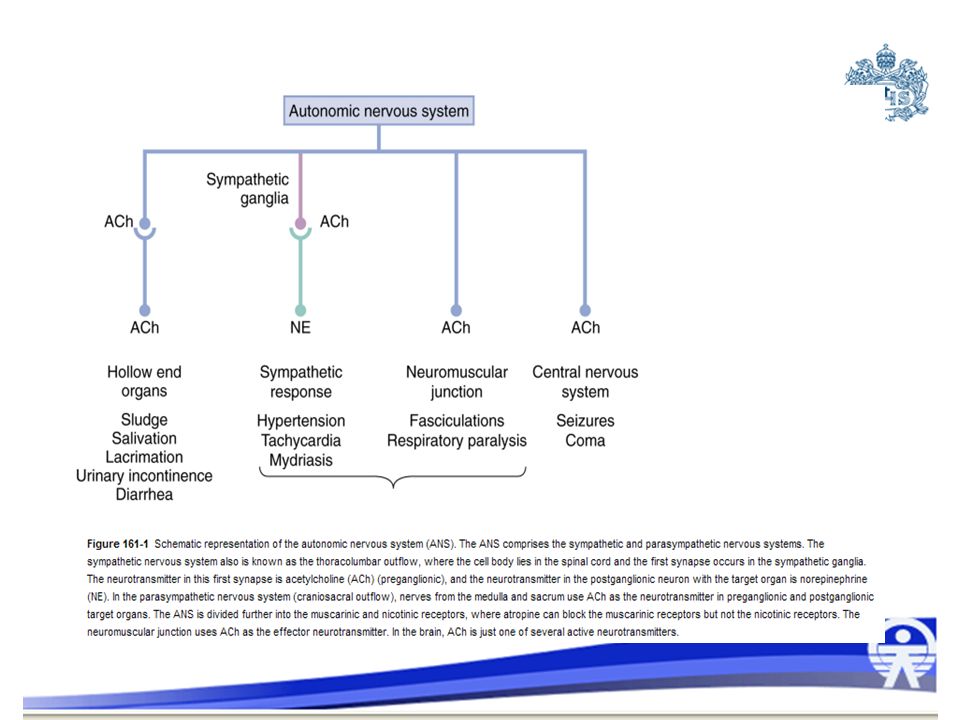


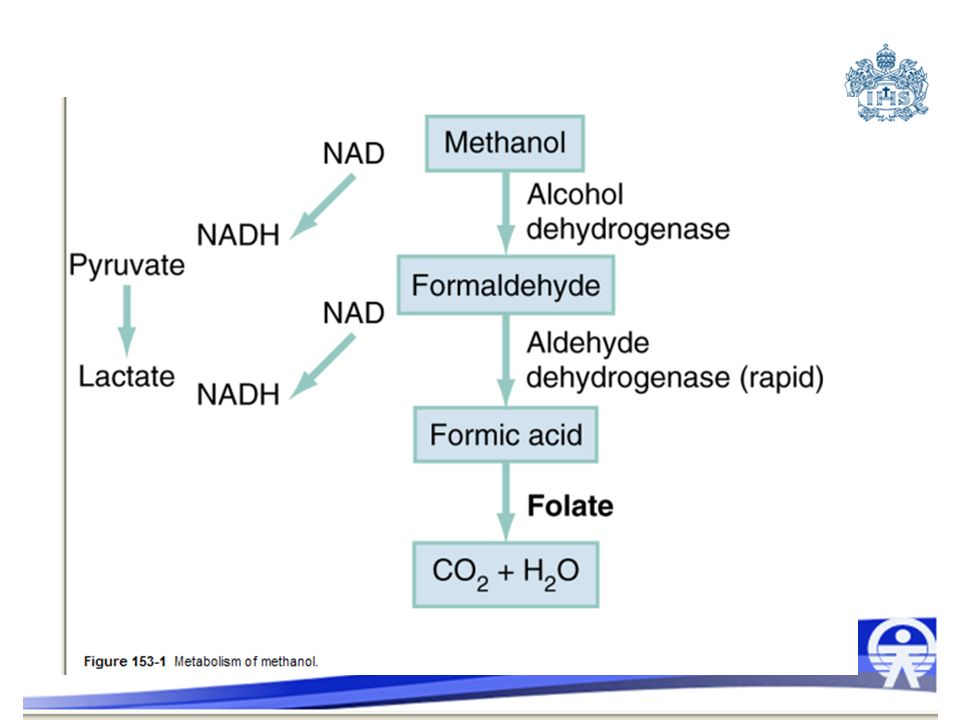




>")









. Protocolo de actuación H.C.Bidasoa>")
Hg+, acumulado por los animales marinos, y incorporado a las cadenas tróficas El dimetil mercurio (CH3)2Hg, el metil mercurio CH3Hg+>")



