Descargar la presentación
La descarga está en progreso. Por favor, espere

1
Dr. Carlos Valdelamar Támara Gineco-Obstetra Universidad de Costa Rica
Amenorrea Dr. Carlos Valdelamar Támara Gineco-Obstetra Universidad de Costa Rica

2
Ausencia de menstruación a los 14 años, y de crecimiento o desarrollo de características sexuales secundarias. Ausencia de menstruación a los 16 años, independientemente de la presencia de un crecimiento normal y del desarrollo de los características sexuales secundarias. En las mujeres que ya han menstruado con anterioridad, la ausencia de menstruación durante un intervalo de tiempo equivalente a por lo menos tres ciclos anteriores o 6 meses. Definición

3
Clasificación tradicional de la amenorrea
secundaria Primaria

4
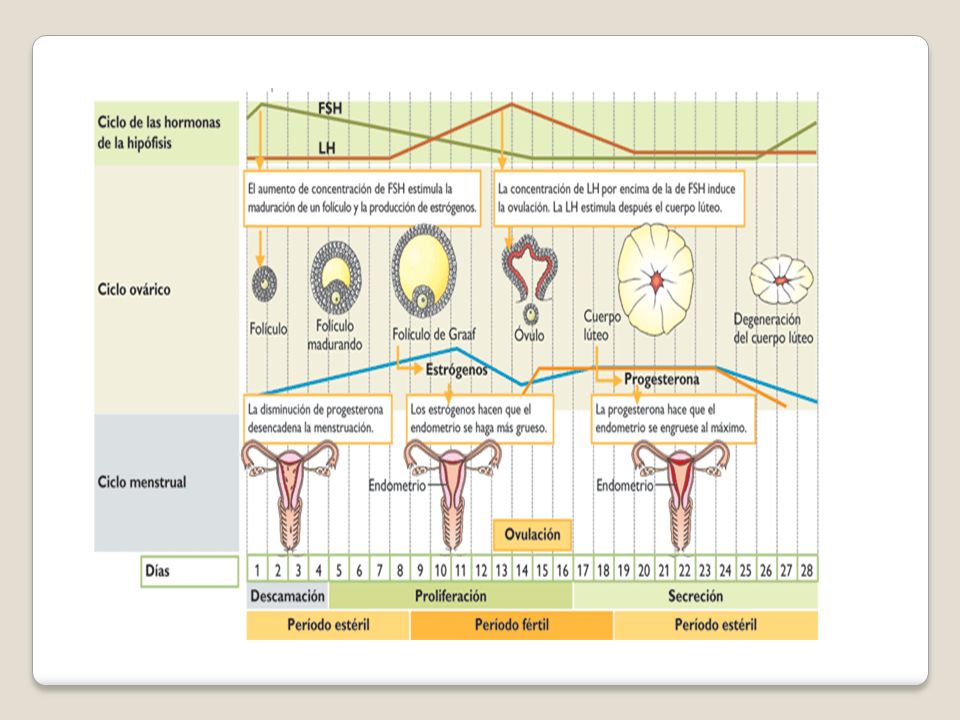
Principios básicos de la función menstrual
Tracto de salida del aparato genital debe estar anatómicamente intacto. El útero también debe contener un endometrio funcional que pueda responder a las hormonas esteroideas sexuales ováricas. Los ovarios deben contener folículos viables que puedan responder a la estimulación de las gonadotrofinas. Función hipotalámica. Principios básicos de la función menstrual

6
Clasificación según el sitio o el nivel del trastorno o la alteración
Trastornos del flujo de salida del aparato genital incluido el útero. Trastornos Ováricos. Trastornos de la adenohipófisis. Trastornos del hipotálamo o del sistema nervioso central

7
Evaluación de la amenorrea: empieza con una anamnesis y una exploración física detalladas. La evaluación recomendada está concebida para separar el sistema reproductor en sus distintos componentes estructurales. Anamnesis: antecedentes menstruales, médicos, de estilo de vida, uso de medicamentos, traumas, signos de estrés físico o psicológico. Exploración física: constitución física (talla, peso, IMC), examen de la piel (textura, coloración, acné, hirsutismo), exploración de las mamas, palpación abdominal, exploración detallada de los genitales externos y del aparato genital inferior.
, examen de la piel (textura, coloración, acné, hirsutismo), exploración de las mamas, palpación abdominal, exploración detallada de los genitales externos y del aparato genital inferior.")
8
Causas especificas de amenorrea

9
Trastornos del tracto de salida del aparato genital y el útero
Como causa de amenorrea, son relativamente poco frecuentes. Las anomalías congénitas del desarrollo de ambos se deben a un fallo en la fusión vertical (himen imperforado, tabique vaginal transverso o atresia cervical) o de una alteración del desarrollo del conducto mülleriano (agenesia mülleriana/vaginal, síndrome de insuficiencia a los andrógenos. Aparecen en la menarquia como amenorrea primaria Los únicos trastornos asociados con una anatomía normal son la estenosis cervical y las adherencias intrauterinas u otras lesiones endometriales causadas por traumatismos quirúrgicos o infecciones. Estos se manifiestan como amenorrea secundaria. Trastornos del tracto de salida del aparato genital y el útero
 o de una alteración del desarrollo del conducto mülleriano (agenesia mülleriana/vaginal, síndrome de insuficiencia a los andrógenos. Aparecen en la menarquia como amenorrea primaria. Los únicos trastornos asociados con una anatomía normal son la estenosis cervical y las adherencias intrauterinas u otras lesiones endometriales causadas por traumatismos quirúrgicos o infecciones. Estos se manifiestan como amenorrea secundaria. Trastornos del tracto de salida del aparato genital y el útero.")
10
Himen imperforado: El himen se forma por la invaginación de la pared posterior del seno urogenital y habitualmente se rompe espontáneamente durante el periodo perinatal. Se observa esporádicamente. Posibilidad de causa genética hereditaria. El examen genital revela un orificio vaginal no visible y una membrana perineal delgada, a menudo protuberante y amoratada en el limite inferior de una masa palpable y fluctuante (hematocolpos). Tabique transverso/atresia cervical: se observa cuando la placa vaginal, formada a partir de los bulbos sinovaginales fusionados, no se rompe ni canaliza durante la embriogénesis. La exploración física revela la presencia de un orificio vaginal normal, una vagina acortada de variadas longitudes, un cuello no visible y un hematocolpos palpable en ele segmento vaginal proximal sobre la obstrucción o una masa pélvica producto de hematómetra y hemosalpinges.

11
La falta de desarrollo de los conductos de Müller es una causa relativamente frecuente de amenorrea primaria, mucho más frecuente que el SIA. Se desconoce la etiología. Puede atribuirse a una mutación activadora en el gen que codifica a la hormona anti-mülleriana o a su receptor, causando exceso de actividad de la hormona. Estas pacientes suelen acudir a la consulta casi al final de la adolescencia o en los primeros años de la edad adulta, bastante después del momento previsto para la menarquia. Agenesia de los conductos de Müller (síndrome de Mayer/Rokytansky-Küster-Hauser

12
Muestran un desarrollo normal y simétrico de las mamas y vello púbico, no presentan vagina visible y no tienen síntomas ni signos de criptomenorrea porque el útero rudimentario no contiene endometrio funcional. El tipo A se caracteriza por un útero simétrico, muscular y rudimentario, y trompas de Falopio normales, y el tipo B, por un útero rudimentario y asimétrico, y trompas de Falopio ausentes o hipoplásicas. Los ovarios son completamente normales, pero uno o ambos pueden no haber descendido, ser hipoplásicos o estar asociados a una hernia inguinal. Las anomalías urológicas son relativamente frecuentes. Suele poder diagnosticarse mediante la anamnesis y la exploración física. Después de la pubertad, una concentración sérica de Testosterona dentro del intervalo normal para la mujer descarta por completo el SIA. Sin embargo, estas pacientes pueden mostrar características similares a las que se observan en algunos tipos de seudohermafroditismo masculino, se justifica un cariotipo, que será definitivo.

13
Síndrome de insensibilidad a los andrógenos (SIA)
El SIA completo (feminización testicular) es una forma de seudohermafroditismo masculino, en ele que se contraponen el sexo gonadal (masculino) con el fenotipo (femenino). Es la tercera causa más frecuente de amenorrea primaria, después de la disgenesia gonadal y la agenesia de los conductos de Müller. Las pacientes con SIA tienen un cariotipo masculino normal (46, XY) y testículos que producen testosterona y hormona anti-mülleriana. En consecuencia, los genitales externos son de una mujer(ausencia de acción de los andrógenos),el cérvix y el útero faltan(a causa de la acción normal de la AMH), y la vagina es corta y tiene un final ciego(derivada sólo del seno urogenital). Síndrome de insensibilidad a los andrógenos (SIA)
 es una forma de seudohermafroditismo masculino, en ele que se contraponen el sexo gonadal (masculino) con el fenotipo (femenino). Es la tercera causa más frecuente de amenorrea primaria, después de la disgenesia gonadal y la agenesia de los conductos de Müller. Las pacientes con SIA tienen un cariotipo masculino normal (46, XY) y testículos que producen testosterona y hormona anti-mülleriana. En consecuencia, los genitales externos son de una mujer(ausencia de acción de los andrógenos),el cérvix y el útero faltan(a causa de la acción normal de la AMH), y la vagina es corta y tiene un final ciego(derivada sólo del seno urogenital). Síndrome de insensibilidad a los andrógenos (SIA)")
14
Son normales al nacer. El crecimiento y desarrollo durante la niñez es por lo general normal. En la pubertad, las mamas se desarrollan por estimulación de los estrógenos derivados de la conversión periférica de niveles elevados de testosterona circulante, sin la oposición de las acciones de los andrógenos. Las mamas pueden tornarse relativamente grandes y presentar anomalías sutiles; al faltar los efectos de la progesterona, tienen un tejido glandular pequeño , pezones de escaso tamaño y aréolas pálidas. Por lo general, los labios menores están subdesarrollados y la vagina es corta y termina de forma ciega. El vello púbico y axilar no se desarrolla a causa de la ausencia de estimulación androgénica. Los testículos pueden ser intraabdominales, pero en ocasiones están parcialmente descendidos; más de la mitad de los pacientes presentan una hernia inguinal SIA

15
El diagnóstico, en general, no es difícil
Las pacientes con SIA completo presentan amenorrea primaria. Su desarrollo sexual secundario es asimétrico(desarrollo de las mamas con ausencia o escaso vello púbico), vagina corta sin cuello visible y sin otros síntomas ni problemas. Al nacer o en la niñez presentan masa o hernia inguinal El diagnóstico, en general, no es difícil
, vagina corta sin cuello visible y sin otros síntomas ni problemas. Al nacer o en la niñez presentan masa o hernia inguinal. El diagnóstico, en general, no es difícil.")
16
La concentración sérica de testosterona permite distinguir fácilmente las pacientes con SIA, porque los valores son normales o moderadamente elevados en comparación con los de varones normales, y mucho mayores que los valores normales observados en otras mujeres. Las concentraciones séricas de LH también están elevadas, lo que refleja insensibilidad a los andrógenos a nivel del hipotálamo y de la hipófisis. Un cariotipo(46, XY) establece firmemente el diagnóstico.
 establece firmemente el diagnóstico..")
17
El SIA completo es la única excepción a la regla de que las gónadas con un cromosoma Y deben extirparse tan pronto como se realice el diagnóstico. SIA incompleta, la sensibilidad es mayor. En consecuencia, el crecimiento de vello púbico puede ir acompañado del desarrollo mamario, y el clítoris puede aumentar de tamaño o puede existir un falo. El tratamiento de las pacientes con SIA completo tiene 2 componentes importantes, uno destinado a la creación de una vagina funcional y otro relacionado con el riesgo de cáncer en los testículos criptorquídeos.

18
Síndrome de Asherman (adherencias intrauterinas)
El diagnóstico se basa principalmente en un elevado índice de sospecha según la anamnesis. Una sangrado escaso o la ausencia tras un tratamiento secuencial con estrógenos exógenos y progestágenos puede ser indicio de una insuficiencia endometrial y corroborar la sospecha clínica. Descrito por Joseph Asherman en 1948 y llamado “amenorrea traumática”, se debe a adherencias intrauterinas que obstruyen o cierran la cavidad uterina, como consecuencia de un traumatismo. El riesgo de estas adherencias intrauterinas aumenta con la inflamación como sucede a causa de una endometritis o de la retención de productos de la concepción, o cuando el endometrio es relativamente fino y está inactivo, como ocurre durante el puerperio.

19
La sonohisterografía o la histerosalpingografía proporciona información específica sobre la ubicación y el tamaño de las adherencias que cierran u obstruyen parcial o totalmente la cavidad endometrial o el canal cervical, mientras que la histeroscopía es definitiva. La histeroscopía quirúrgica es el principal método para el tratamiento de las adherencias intrauterinas. Se recomienda tratamiento con altas dosis de estrógenos exógenos por 4 semanas posterior a la cirugía, para promover la nueva epitelización y proliferación endometrial, seguido con un progestágeno en la semana final.

20
La estenosis cervical grave con obstrucción completa del flujo de salida es una complicación poco frecuente de los procedimientos de conización cervical o de otros tratamientos quirúrgicos para NIC. Dismenorrea. Producción de una ligera pérdida prolongada después de la menstruación son los más comunes. La amenorrea es poco frecuente. El tratamiento consiste en la dilatación cuidadosa del cérvix. Estenosis cervical

21
Trastornos ováricos Anovulación crónica e insuficiencia ovárica.

22
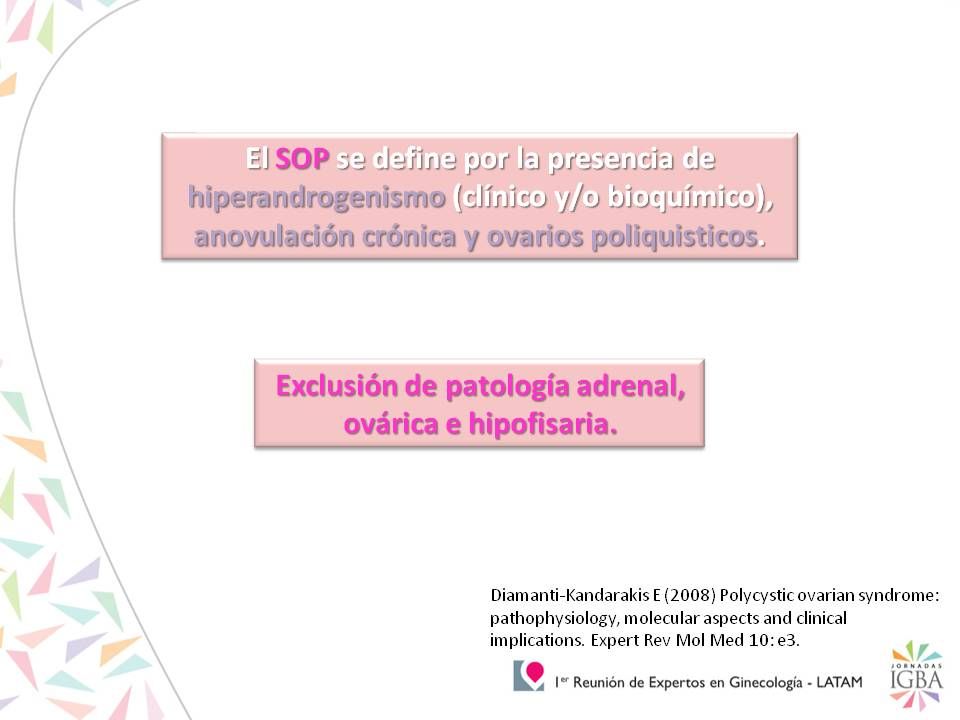
Poliquistosis ovárica
Hiperprolactinemia Trastornos tiroideos Anovulación crónica

23
Hipotiroidismo e hipertiroidismo primario ambos pueden causar anovulación crónica y amenorrea.
El restablecimiento de los niveles normales de hormona tiroidea provoca el retorno de los ciclos ovulatorios. Algunas mujeres con hipotiroidismo presentan Hiperprolactinemia secundaria e incluso galactorrea. Las pacientes con hipotiroidismo primario e HPRL pueden presentar amenorrea primaria y secundaria Trastornos tiroideos.

24
La HPRL es una de las causas más comunes de amenorrea secundaria y, se aparece antes de la menarquia, puede causar también retraso de la pubertad y amenorrea primaria. Está justificada la medición de la concentración sérica de la prolactina en todas las mujeres con amenorrea. Solo un tercio de las mujeres con HPRL presentan galactorrea, probablemente porque la producción de leche requiere estrógenos. Lesiones que afecten los dermatomas que incluyen la mama pueden activar vías neurosensitivas aferentes que estimula la secreción de prolactina similar a la succión. Para evitar los estudios de imágenes innecesarios y costosos, antes de diagnosticar una HPRL es mejor repetir y confirmar los valores ligeramente elevados de prolactina. HPRL produce anovulación y amenorrea está relacionada con la interrupción e inhibición de la secreción pulsátil normal de GnRH por hipotálamo, que produce niveles ineficaces o francamente bajos de secreción de gonadotrofinas. Causas medicamentosas: anfetaminas, benzodiazepinas,buritofenonas, metoclopramida, metil-dopa, opiáceos, fenotiacinas, reserpina y antidepresivos tricíclicos. Hiperprolactinemia

25
Es una Alteración Endócrina Frecuente que se observa asociada a:
Alteraciones del Ciclo Menstrual 20 % Trastornos de la Fertilidad 40 % Amenorrea y Galactorrea 80 % Adenomas de Hipófisis %

26
CAUSAS NO HIPOFISARIAS
FISIOLÓGICA Embarazo Lactancia Sueño REFLEJA Traumatismos pared torácica (cirugía – herpes zóster – quemaduras) Succión del pezón CAUSAS NO HIPOFISARIAS Estrés Poliquistosis ovárica Hipotiroidismo Insuf. renal crónica TUMORAL Tumores hipofisiarios Prolactinomas: Microadenomas Macroadenomas Otros adenomas Enf. hipotalámica y del tallo hipofisiario Enf. granulomatosa Cráneo faringeoma Sind. silla turca vacía Hipofisitis autoinmune Tumores productores de estrógenos TODO COMPLETITO EL EJE Y DESPUES LA ANIMACIÓN DE SINTESOFT
 Succión del pezón. CAUSAS NO HIPOFISARIAS. Estrés. Poliquistosis ovárica. Hipotiroidismo. Insuf. renal crónica. TUMORAL. Tumores hipofisiarios. Prolactinomas: Microadenomas. Macroadenomas. Otros adenomas. Enf. hipotalámica y del tallo hipofisiario. Enf. granulomatosa. Cráneo faringeoma. Sind. silla turca vacía. Hipofisitis autoinmune. Tumores productores de estrógenos. TODO COMPLETITO EL EJE Y DESPUES LA ANIMACIÓN DE SINTESOFT.")
27
MEDICAMENTOSA Depleción de las reservas de dopamina Reserpina
Bloqueo de la fijación al receptor de dopamina Fenotiacinas Butirofenonas Benzodiacepinas Metoclopramida Domperidona Sulpiride Interferencia con la síntesis de dopamina Alfa metil dopa Inhibición de la liberación de dopamina Opiáceos Bloqueo de la fijación al receptor H2 Cimetidina Ranitidina Difenhidramina Bloqueadores de los canales del calcio Verapamil Mecanismo desconocido o mixto Antidepresivos tricíclicos Derivados de la papaverina Estrógenos

28
¿Por qué consultan los pacientes?
Alteración del ciclo (oligomenorreas) 20% Infertilidad (anovulación) 40% Amenorrea Galactorrea Galactorrea + Amenorrea 80% Signos de androgenización de la libido Impotencia
 20% Infertilidad (anovulación) 40% Amenorrea. Galactorrea. Galactorrea + Amenorrea 80% Signos de androgenización. de la libido. Impotencia.")
29
PROLACTINOMAS Los Prolactinomas constituyen los adenomas pituitarios hormono-secretantes más frecuentes Constituyen el 40% de los adenomas hipofisarios en adultos Es más habitual en mujeres entre los 20 y 50 años. Microadenomas menor a 10mm Macroadenomas mayor a 10mm

30
Instituto Ginecológico Buenos Aires
PROLACTINOMAS Dra.Claudia Peyrallo Instituto Ginecológico Buenos Aires IGBA - Argentina

31
Síndrome de ovarios poliquísticos: La endocrinopatía de la edad fértil

39
Se define como la formación incompleta o defectuosa de las gónadas a causa de la alteración en la migración o la organización de las células germinativas, debido a anomalías estructurales o numéricas en los cromosomas sexuales o mutaciones en los genes que participan en la formación de la cresta urogenital y en la diferenciación sexual de las gónadas bipotenciales. Se encuentra entre las causas más frecuentes de amenorrea primaria (30-40%). Dada la ausencia de folículos ováricos o su rápida depleción durante la embriogénesis o los primero años de vida, las gónadas solo contienen estroma y aparecen como bandas fibrosas. Disgenesia Gonadal
. Dada la ausencia de folículos ováricos o su rápida depleción durante la embriogénesis o los primero años de vida, las gónadas solo contienen estroma y aparecen como bandas fibrosas. Disgenesia Gonadal.")
40
La mayoría de las pacientes tienen una anomalía evidente que afecta a un cromosoma X.
25% cariotipo 46,XX normal, y pueden contener una anomalía más sutil que afecta a uno o más genes específicos en el cromosoma X, necesarios para la función ovárica normal, algunas mujeres 46,XX presentan sordera neurosensitiva (síndrome Perrault). Sin duda la forma más frecuente es el síndrome de Turner. Disgenesia Gonadal
. Sin duda la forma más frecuente es el síndrome de Turner. Disgenesia Gonadal.")
42
Síndrome de Turner En el año 1938 Henry Turner describió por primera vez en la revista Endocrinology, un grupo de siete mujeres con edades comprendidas entre los 15 y los 23 años, que presentaban una serie de alteraciones físicas que llamaron su atención y que hizo que las agrupara en un nuevo síndrome: EL SÍNDROME DE TURNER.

43
Trastorno bien conocido y cuidadosamente estudiado, que clásicamente se asocia a un cariotipo 45,X.
El fenotipo clásico incluye talla baja, ausencia de desarrollo sexual, cuello corto, implantación baja de las orejas y de la línea posterior del cuello, pezones muy separados (tórax en escudo), cuartos metacarpianos cortos y aumento del ángulo del codo (cúbito valgo). Las pruebas indican que el fenotipo especifico se relacionan, en parte con el origen parenteral del cromosoma X; la mayoría de los que presentan cariotipo 45,X mantienen el cromosoma X materno. Síndrome de Turner
, cuartos metacarpianos cortos y aumento del ángulo del codo (cúbito valgo). Las pruebas indican que el fenotipo especifico se relacionan, en parte con el origen parenteral del cromosoma X; la mayoría de los que presentan cariotipo 45,X mantienen el cromosoma X materno. Síndrome de Turner.")
44
Si durante la niñez no se reconocen por el fenotipo o el escaso crecimiento, presentan al momento de la pubertad amenorrea y ausencia del desarrollo sexual secundario. Por lo general el diagnóstico se realiza fácilmente, basándose en el fenotipo y la detección del hipogonadismo hipergonadótropo. El cariotipo es definitivo. Síndrome de Turner

45
Amplia variedad de problemas médicos con implicaciones para la salud, tanto o más importante que las asociadas al hipogonadismo y sus consecuencias directas. 1/3 presenta anomalías cardiovasculares, como válvula aórtica bicúspide, coartación de la aorta, prolapso de la válvula mitral y aneurisma aórtico. Anomalías renales: Riñón en herradura, agenesia renal unilateral o riñón pélvico, anomalías de rotación, y duplicación parcial o completa del sistema colector. Trastornos auto inmunitarios: tiroiditis, diabetes tipo 1, hepatitis auto inmunitarias y trombocitopenia, celiaquía. La hipoacusia también es frecuente. Síndrome de Turner

46
1 vez si es normal, c/3-5 años si es anómala
Ecocardiografía Al diagnóstico, al menos una vez entre los años, c/5 años si es normal, > frecuencia si está alterada. Ecografía Renal 1 vez si es normal, c/3-5 años si es anómala Hemograma completo, glicemia ayunas, perfil lipídico, PFR y enzimas hepáticas cada 2 años TSH y T4 libre (en el momento del diagnóstico y cada 1-2 años) Anticuerpos antiendomisio, para detectar la celiaquía Audiometría, al momento del diagnóstico, al menos una vez en la adolescencia o iniciada la edad adulta y cada 10 años si es normal
 Anticuerpos antiendomisio, para detectar la celiaquía. Audiometría, al momento del diagnóstico, al menos una vez en la adolescencia o iniciada la edad adulta y cada 10 años si es normal.")
47
El promedio del rendimiento intelectual se encuentra dentro de los limites normales, aunque la prevalencia del déficit de atención con hiperactividad es mayor. La mortalidad total es 3 veces mayor y se relaciona principalmente con enfermedades circulatorias(HTA), diabetes, hepatopatías y nefropatías. El riesgo total de cáncer es similar al de la población general, pero la incidencia de tumores del SNC, cáncer vesical y endometrial puede ser más alta. El de mama es menor.
, diabetes, hepatopatías y nefropatías. El riesgo total de cáncer es similar al de la población general, pero la incidencia de tumores del SNC, cáncer vesical y endometrial puede ser más alta. El de mama es menor.")
48
El tratamiento con somatotropina (GH), permite alcanzar una estatura final superior a 150 cm.
El tratamiento con Estrógenos debe administrarse en el momento adecuado, con el objetivo de minimizar los efectos adversos sobre el crecimiento y la talla adulta, e inducir la pubertad en una edad aproximadamente normal. Preferentemente, debe comenzar antes de los 15 años, pero después de los 12 años, cuando el crecimiento es prioritario, salgo que ya se haya alcanzado la estatura máxima posible La donación de ovocitos ofrece la posibilidad de un embarazo, pero la demanda cardiovasculares del embarazo plantean riesgos característicos y posiblemente graves que se deben considerar cuidadosamente. El riesgo de muerte durante el embarazo está aumentado 100 veces, debido principalmente a las complicaciones de la disección o rotura de la aorta.

49
Forma de disgenesia gonadal claramente diferente y menos frecuente, caracterizada por el cariotipo 46,XY. Fenotipo femenino a pesar del cromosoma Y porque las gónadas disgenéticas no producen AMH ni andrógenos. Por lo tanto habrá vagina, cuello y trompas de Falopio y no hay masculinización de genitales. Presentan retraso de la maduración sexual y amenorrea primaria. Se debe realizar gonadectomía por el importante riesgo de transformación maligna de elementos testiculares ocultos (20- 30%) Muestran crecimiento y desarrollo intelectual normales, sin prevalencia de problema médico especifico, y no requieren evaluaciones ni tratamientos específicos más allá de los relacionados con el tratamiento hormonal destinado a inducir la maduración sexual. Síndrome de Swyer
 Muestran crecimiento y desarrollo intelectual normales, sin prevalencia de problema médico especifico, y no requieren evaluaciones ni tratamientos específicos más allá de los relacionados con el tratamiento hormonal destinado a inducir la maduración sexual. Síndrome de Swyer.")
50
Disgenesia Gonadal 46, XX Algunas mujeres con amenorrea primaria y disgenesia gonadal (gónadas en cordón) tienen un cariotipo normal 46,XX, lo que constituye una prueba indirecta de que los genes autosómicos también tienen un papel importante en la diferenciación ovárica. Las mujeres afectadas tienen una talla normal y en la mayoría de los casos, no presentan anomalías somáticas evidentes.
 tienen un cariotipo normal 46,XX, lo que constituye una prueba indirecta de que los genes autosómicos también tienen un papel importante en la diferenciación ovárica. Las mujeres afectadas tienen una talla normal y en la mayoría de los casos, no presentan anomalías somáticas evidentes.")
51
Insuficiencia ovárica prematura
El fallo ovárico prematuro, tradicionalmente definido como la presencia de hipogonadismo hipergonadótropo y amenorrea antes de los 40 años, es un trastorno heterogéneo que varía ampliamente en cuanto a causas y fenotipos. Caracterizada por la continua disminución de la función ovárica. Muchas mujeres presentan función ovárica y ovulación intermitente. 5-10% puede concebir y llevar a término el embarazo. Insuficiencia ovárica prematura

52
Causa amenorrea secundaria poco después de la pubertad, pero también puede presentarse en cualquier momento antes de la menarquia; se diferencia de la disgenesia gonadal por la morfología e histología ováricas que, en lugar de gónadas en cordón, presenta ovarios semejantes a los de mujeres posmenopáusicas. El 1% de las mujeres pueden presentarla antes de los 40 años. Entre las causas conocidas importantes de insuficiencia ovárica prematura, se encuentran las anomalías cromosómicas numéricas y estructurales, las premutaciones en el cromosoma X frágil, los trastornos autoinmunitarios, la radioterapia y la quimioterapia.

53
Anomalías cromosómicas numéricas y estructurales:
La revisión de los cariotipos obtenidos en mujeres con amenorrea secundaria revela la variedad de posibilidades y demuestra la importancia de realizar un cariotipo en mujeres IOP. La ½ de las anomalías observadas fueron numéricas, afectando el mosaicismo del cromosoma X (como las extirpes celulares 45X, 46XX, 47XXX) o el mosaicismo del cromosoma Y(46XY, 47XYY y 47XY); las restantes incluyen una variedad de translocaciones, delecciones, y otras anomalías estructurales del cromosoma X, e incluso algunas con un cariotipo puro 46,XY. Algunas presentan delecciones y translocaciones que afectan el brazo corto o largo del cromosoma X. Solo la ½ de las que tienen delecciones en Xp, presentan amenorrea primaria y disgenesia gonadal; las restantes menstrúan y con frecuencia presentan IOP.
 o el mosaicismo del cromosoma Y(46XY, 47XYY y 47XY); las restantes incluyen una variedad de translocaciones, delecciones, y otras anomalías estructurales del cromosoma X, e incluso algunas con un cariotipo puro 46,XY. Algunas presentan delecciones y translocaciones que afectan el brazo corto o largo del cromosoma X. Solo la ½ de las que tienen delecciones en Xp, presentan amenorrea primaria y disgenesia gonadal; las restantes menstrúan y con frecuencia presentan IOP.")
54
Premutaciones en el cromosoma X frágil
El síndrome del cromosoma X frágil es causado por un cambio en un gen llamado FMR1. Una pequeña parte del código del gen se repite en un área frágil del cromosoma X. Cuantas más repeticiones se presenten, mayor será la probabilidad de que haya un problema. El gen FMR1 produce una proteína que se necesita para que el cerebro crezca apropiadamente. Un defecto en este gen hace que el cuerpo produzca muy poco de esta proteína o nada en absoluto. Tanto los niños como las niñas pueden resultar afectados, pero debido a que los niños tienen únicamente un cromosoma X, es más probable que un solo cromosoma X frágil los afecte con más gravedad. Usted puede tener el síndrome del cromosoma X frágil incluso si sus padres no lo tienen. Puede que se presenten un antecedente familiar del síndrome del cromosoma X frágil, problemas del desarrollo o discapacidad intelectual.

55
Los problemas de comportamiento asociados con el síndrome del cromosoma X frágil abarcan:
Retraso para gatear, caminar o voltearse Palmotear o morderse las manos Comportamiento hiperactivo o impulsivo Discapacidad intelectual Retraso en el habla y el lenguaje Tendencia a evitar el contacto visual Los signos físicos pueden abarcar: Pies llanos Articulaciones flexibles y tono muscular bajo Tamaño del cuerpo grande Orejas o frente grandes con una mandíbula prominente Cara larga Piel suave Algunos de estos problemas están presentes en el nacimiento, mientras que es posible que otros no se presenten hasta después de la pubertad.

56
Premutaciones en el cromosoma X frágil
Un grupo de trastornos clínicamente importantes entre ellos la IOP, conlleva a una mutación dinámica de la secuencia de repetición de trinucleótidos (CGG) en el gen FMR1,localizado cerca el extremo terminal del brazo largo del cromosoma X. La forma completamente expandida de la mutación, caracterizada por más de 200 repeticiones CGG, causa el síndrome del cromosoma X frágil (FXS), la causa genética conocida más frecuente de retraso mental y autismo. Premutaciones en el cromosoma X frágil
 en el gen FMR1,localizado cerca el extremo terminal del brazo largo del cromosoma X. La forma completamente expandida de la mutación, caracterizada por más de 200 repeticiones CGG, causa el síndrome del cromosoma X frágil (FXS), la causa genética conocida más frecuente de retraso mental y autismo. Premutaciones en el cromosoma X frágil.")
57
Premutaciones en el cromosoma X frágil
La premutación, con repeticiones, está asociada a dos trastornos diferentes de FXS. Uno es el síndrome de temblor/ataxia, afecta principalmente a hombres. El otro es la IOP afecta al 15% de las portadoras de la premutación Las portadoras a menudo presentan signos de envejecimiento reproductor prematuro. La duración de los ciclos y de la fase folicular es más corta. La menopausia se presenta 5 años antes que el promedio. Premutaciones en el cromosoma X frágil

59
Trastornos autoinmunitarios
Una prueba (+) para anticuerpos antisuprarrenales o anti-21-hidroxilasa es suficiente para establecer el diagnóstico. La biopsia ovárica es innecesaria y no recomendable La estrecha asociación entre la insuficiencia suprarrenal y la IO autoinmunitarias justifica el cribado para la detección de anticuerpos antisuprarrenales en todas las mujeres con IOP en el momento del diagnóstico. Trastornos autoinmunitarios
 para anticuerpos antisuprarrenales o anti-21-hidroxilasa es suficiente para establecer el diagnóstico. La biopsia ovárica es innecesaria y no recomendable. La estrecha asociación entre la insuficiencia suprarrenal y la IO autoinmunitarias justifica el cribado para la detección de anticuerpos antisuprarrenales en todas las mujeres con IOP en el momento del diagnóstico. Trastornos autoinmunitarios.")
60
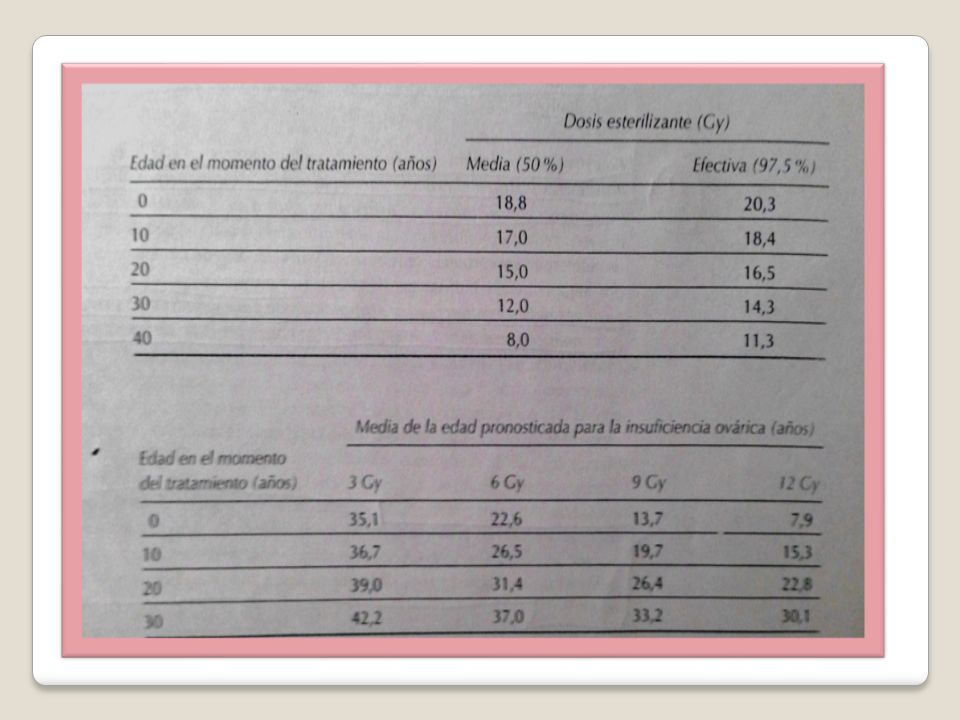
Los efectos adversos de la radiación sobre el ovario depende de la edad de la paciente, la dosis de radiación y el campo a radiar. En mujeres jóvenes puede causar una amenorrea transitoria que desaparece en meses. Las que reciben yodo radioactivo presentan suspensión transitoria del ciclo ovárico normal. Algunas sufrirán insuficiencia ovárica inmediata e irreversible, e incluso las que la recuperan pueden presentar más tarde envejecimiento ovárico y menopausia prematuras. Los ovarios de las mujeres de más edad son más sensibles a los efectos de la radiación. Radioterapia

62
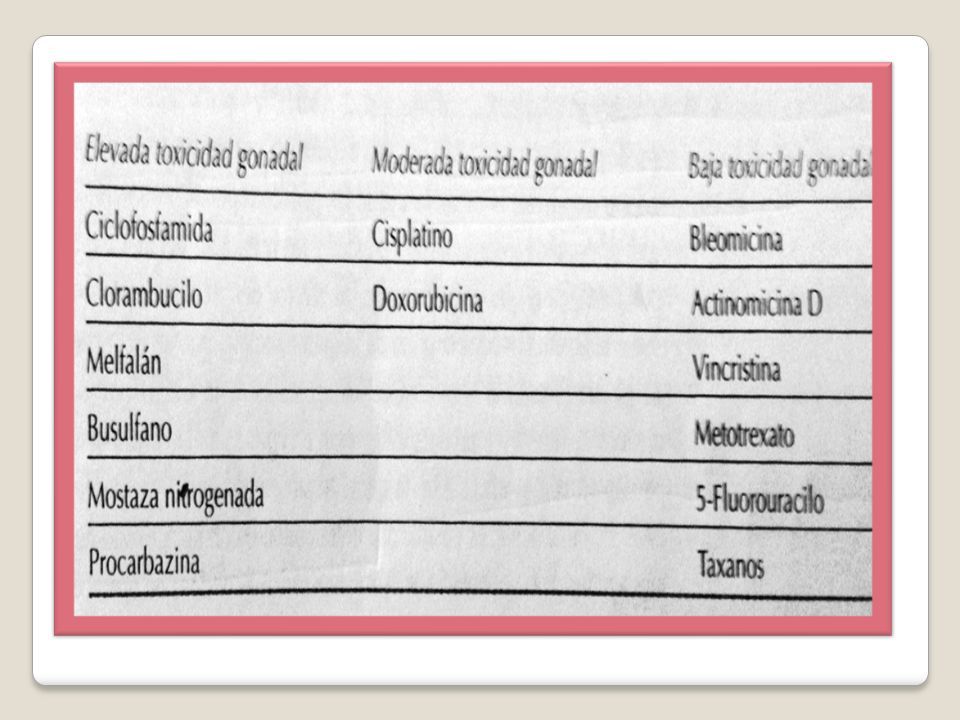
La mayoría de los antineoplásicos actúan sobres las células en división y, por lo tanto, es probable que no tengan efectos adversos significativos sobre los ovocitos, aunque hay muchos que lo tienen. De hecho, la provisión fija de ovocitos es extremadamente sensible a los fármacos citotóxicos. La quimioterapia causa una disminución de la reserva de folículos primordiales que depende del fármaco y de la dosis y es una de las causas relativamente frecuente de IOP. Quimioterapia

65
Es importante destacar que el tratamiento eficaz de la IOP necesita asesoramiento y respaldo emocional profundos, evaluación específica y tratamiento farmacológico. Tratamiento hormonal: sin reposición estrogénica exógena existe riesgo de osteopenia y osteoporosis, así de cardiopatías isquémicas tempranas. Síntomas de deficiencia estrogénica como crisis vasomotoras y atrofia genitourinaria. Ejercicio, dieta, calcio y vitamina D. Tratamiento de IOP

67
Fig. 2: Enfoque de la figura anterior en la mitad del encéfalo.
Fig. 6: RNM. Corte por la mitad de la cabeza. La glándula hipofisaria se encuentra en el interior de la Silla Turca. La neurohipófisis se observa más blanca (híper-intensa) que la adenohipófisis. Fig. 2: Enfoque de la figura anterior en la mitad del encéfalo.
 que la adenohipófisis. Fig. 2: Enfoque de la figura anterior en la mitad del encéfalo.")
68
Síndrome de la silla vacía
Es un nombre poco apropiado porque, la silla turca no está vacía sino aumentada de tamaño, aunque aparezca vacía en las imágenes porque contiene LCR, incluso dentro del espacio subaracnoideo pero extendiéndose hacia abajo en la fosa hipofisiaria. Se incluye este trastorno, en la exposición de los adenomas hipofisiarios, porque en la mayoría de los casos es el resultado de la extirpación o destrucción previa de una adenoma mediante cirugía, radiación o infarto. También puede deberse a un defecto congénito localizado en el diafragma de la silla turca (silla turca vacía primaria). El tejido hipofisiario remanente, normal, está aplanado contra el suelo, que puede desmineralizarse a causa del aumento de la presión dentro de la fosa hipofisiaria. Síndrome de la silla vacía
. El tejido hipofisiario remanente, normal, está aplanado contra el suelo, que puede desmineralizarse a causa del aumento de la presión dentro de la fosa hipofisiaria. Síndrome de la silla vacía.")
69
Fig1: RNM. Corte por la mitad de la cabeza
Fig1: RNM. Corte por la mitad de la cabeza. Ampliación de la región de la Silla Turca. Fig. 2: Detalle de la ampliación anterior: Encima de la silla se ve la sección del Quiasma Óptico (flecha amarilla). La flecha roja señala el Tallo de la Hipófisis acodado sobre la pared posterior de la Silla. El fondo de la Silla está almohadillado por la Hipófisis aplastada (flecha verde). Fig. 3: RNM en la secuencia en la que el Líquido Cefalorraquídeo aparece blanco. Corte transversal de la cabeza del mismo paciente que las figuras anteriores. La Silla Turca se ve rellena de líquido y, en su fondo, la Hipófisis aplastada (flecha roja). El punto amarillo señala la sección del Quiasma Óptico.
. La flecha roja señala el Tallo de la Hipófisis acodado sobre la pared posterior de la Silla. El fondo de la Silla está almohadillado por la Hipófisis aplastada (flecha verde). Fig. 3: RNM en la secuencia en la que el Líquido Cefalorraquídeo aparece blanco. Corte transversal de la cabeza del mismo paciente que las figuras anteriores. La Silla Turca se ve rellena de líquido y, en su fondo, la Hipófisis aplastada (flecha roja). El punto amarillo señala la sección del Quiasma Óptico.")
70
Síndrome de Sheehan

71
Lesiones hipofisarias infiltrantes
Hemocromatosis Hipofisitis Linfocítica Trastorno autosómico recesivo, mutación en el cromosoma 6. Sobrecarga férrica en el parénquima y daño tisular. Causa hipogonadismo hipogonadótropico. La forma adquirida por múltiples transfusiones. Trastorno autoinmunitario poco común. Aumenta su tamaño. Embarazo o 6 meses posparto. Destrucción puntual o difusa de la adenohipófisis y fibrosis. Lesiones hipofisarias infiltrantes

72
Trastornos de la función hipotalámica
Amenorrea hipotalámica: el diagnóstico se realiza por exclusión, basándose en la detección de una concentración sérica de FSH baja o normal, a pesar de los bajos niveles de estrógenos, sin lesión tumoral en la silla turca ni razón para sospechar la existencia de otras causas hipofisarias poco frecuentes de hipogonadismo hipogonadótropo. se asocia con estrés físico, nutricional o emocional, lo que sugiere que representa una inhibición funcional de la reproducción como respuesta psicobiológica a acontecimientos de la vida. Trastornos de la función hipotalámica

73
Trastornos de la conducta alimentaria
En la anorexia, la amenorrea precede a la pérdida de peso que comienza con la dieta y a una restricción especifica de las grasas. La osteopenia y la osteoporosis se encuentran entre las complicaciones más graves. En la bulimia tienen menstruaciones irregulares, pero no amenorrea Santa Wilgefortis o Santa Librada.

74
Habitualmente, los ciclos previamente normales se vuelven irregulares después de iniciar ejercicio y progresan hasta la amenorrea a medida que su intensidad aumenta cuando van acompañadas de la pérdida de peso. Parece que un equilibrio energético negativo, que se produce cuando el gasto energía excede la energía disponible (derivada de la ingesta y de los depósitos disponibles), predispone a una interrupción pulsátil de gonadotrofinas y a una pérdida de la función menstrual. Ejercicio y amenorrea
, predispone a una interrupción pulsátil de gonadotrofinas y a una pérdida de la función menstrual. Ejercicio y amenorrea.")
75
Deficiencia congénita de GnRH, se asocia con anosmia o hiposmia (ausencia o deterioro del sentido del olfato) Trastornos ligado al cromosoma X. En la pubertad las niñas y los niños presentan, generalmente, un retraso del crecimiento y del desarrollo sexual Síndrome de Kallmann

Presentaciones similares