Descargar la presentación
La descarga está en progreso. Por favor, espere

1
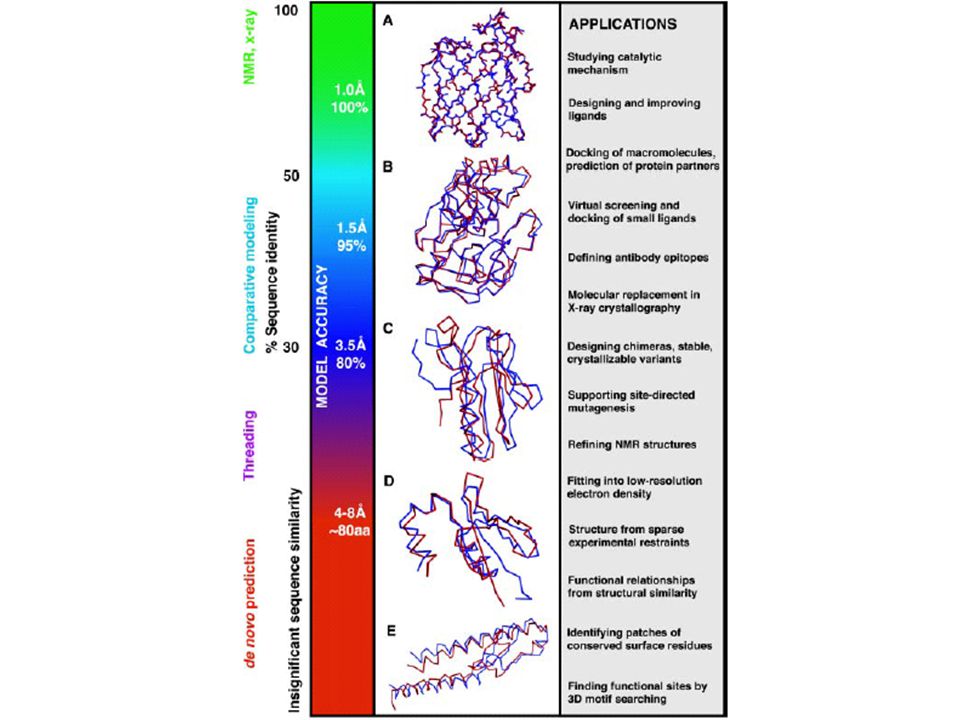
En PDB hay 20 000 estructuras experimentales de proteínas.
En Swiss-Prot y TrEMBL hay secuencias protéicas. La información estructural no es de fácil acceso en muchas proteínas. Gran demanda por conocer la estructura con metodos computacionales que predicen la estructura protéica.

2
¿Porqué modelamos proteínas?
Por ser “fácil”. Por lo difícil y trabajoso que es NMR y Rayos X. Porque puede generar modelos que tienen exactitud y calidad comparable con estructuras generadas por NMR y Rayos X.

4
MODELAMIENTO DE PROTEÍNAS POR HOMOLOGÍA

5
estrategias basadas en información de secuencia,
Consiste en seleccionar homólogos por alineamiento estructural del banco de datos de secuencias usando: estrategias basadas en información de secuencia, información funcional, diferentes tipos de matrices de mutaciones. La construcción del modelo requiere por lo menos de un patrón (template) cuya secuencia sea significativamente similar a la secuencia problema.
 cuya secuencia sea significativamente similar a la secuencia problema.")
6
PASOS A SEGUIR... Reconocimiento de un TEMPLATE. Alineamiento
Secuencias Estructura Construcción del modelo Backbone Loops Cadenas laterales Refinamiento Evaluación

8
1) Selección del Template
Se utiliza la librería Protein Data Bank. Si el template tiene regiones distintas del target, el proceso del modelo se cortaría en bloques independientes y separados. No se utilizan los modelos que sólo presentan los C, por tener una calidad estructural pobre. Se utilizan files: Probable estructura cuaternaria Indicadores empiricos de energía force field ANOLEA

11
2) Alineamiento Debe caer en la zona segura de modelamiento por homología; el porcentaje de identidad entre las secuencias se determina con un simple alineamiento usando programas como BLAST o FASTA.

12
Es muy dificil alinear dos secuencias que presentan identidad pobre, en este caso se utilizan otros homólogos. Generalmente se usan 5 templates y se alinean por bloque (algoritmo de mínimos cuadrados). Se calcula un alineamiento local de las secuencia target con el principal template seguido de un método heurístico para mejorar el alineamiento; tambien se optimizan inserciones y deleciones tomando como base la estructura del template.
. Se calcula un alineamiento local de las secuencia target con el principal template seguido de un método heurístico para mejorar el alineamiento; tambien se optimizan inserciones y deleciones tomando como base la estructura del template.")
14
3) Construyendo el modelo
Backbone Loops Cadenas laterales Refinar el modelo

15
BACKBONE Para generar el núcleo del modelo, la posicion de los átomos del backbone son promediados en la estructura del template. Se observa la similitud con el target. Las coordenadas del template no se usan en las regiones de deleciones o inserciones generadas en el alienamiento. Para esta parte se ensamblan segmentos compatibles con los constructos vecinos usando en programa CSP.

17
LOOPS El mejor loop es seleccionado usando scores con force field, interacciones con formación de puentes de hidrógenos y los impedimentos estéricos. Si no hay un loop aceptable se flanquean los residuos y son incluidos en un fragmento reconstruido con mayor flexibilidad. También se utiliza una librería de loops de estructuras experimentales para encontrar el más compatible.

19
Cadenas laterales La reconstrucción se basa en la posición de las cargas de los residuos en su correspondiente template, se inicia por residuos conservados. Las posibles conformaciones de las cadenas laterales con selecionadas de la librería de rotámeros. Para seleccionar la conformación más apropiada se ven los scores de interacciones favorables y los no favorables.

21
Los residuos de superficie, tienen un porcentaje mucho menor (50%).
En residuos de zonas del núcleo hidrofóbico, la predicción de exactitud debe ser alta (90%). Los residuos de superficie, tienen un porcentaje mucho menor (50%). RAZONES: La superficie tiene cadenas más flexibles y adoptan múltiples conformaciones. Las funciones de energía usadas en los scores de rotámeros son fácilmente obtenidas en zonas hidrofóbicas; en superficie las interacciones electrostáticas son menos exactas, que incluyen los ptes. de hidrógeno que formaría con el agua.
. Los residuos de superficie, tienen un porcentaje mucho menor (50%). RAZONES: La superficie tiene cadenas más flexibles y adoptan múltiples conformaciones. Las funciones de energía usadas en los scores de rotámeros son fácilmente obtenidas en zonas hidrofóbicas; en superficie las interacciones electrostáticas son menos exactas, que incluyen los ptes. de hidrógeno que formaría con el agua.")
22
Refinar el modelo: Minimización de Energía
Al colocar cadenas laterales, se debe corregir el backbone. GROMOS96 es el force field que evalúa las desviaciones introducidas por al algoritmo de modelamiento, es por esto que se realiza una minimización de energía; se evalúan los errores conformacionales del modelo.

23
4) EVALUACIÓN La exactitud del modelo puede variar significativamente dentro de diferentes regiones de la misma proteína: las regiones altamente conservadas del núcleo son mucho mas confiables que los loops variables o la superficie de la proteína.

24
Los errores de la proteina modelada depende de:
Porcentaje de identidad en secuencia entre el template y el target: si es mas de 90% ; entre 50 y 90% es aceptable (presenta errores locales); menos del 25% el alineamiento es el gran problema. Número de errores en el template
; menos del 25% el alineamiento es el gran problema. Número de errores en el template.")
25
Para estimar los errores, hay dos caminos diferentes:
Calcular la energía basandose en force fiels, chequea si el tamaño de uniones y los ángulos de unión están en un rango aceptable. Determinar los índices de normalidad que describen que tanto se asemeja las características del modelo con las características de la estructura real. Basado en análisis de distancias interatómicas y sus contactos

26
En Swiss Model: Para evaluar el resultado de “first approach” se puede utilizar DeepView, ya que permite ajustes manuales del alineamiento que se verifican visualemtne. Para modificar proyectos, se debe volver a someterlo al servidor.

28
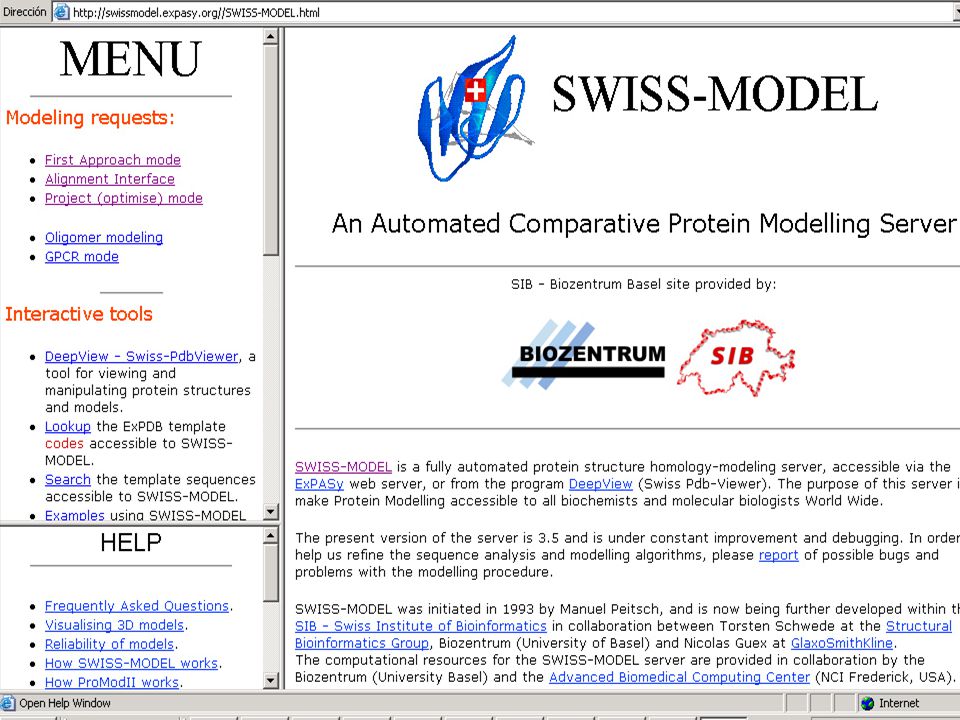
SWISS-MODEL

30
First approach mode Se requiere sólo de la secuencia de aminoácidos como dato. El servidor busca automáticamente los posibles templates, opcionalmente se puede especificar 5 templates, de ExPDB o de propios files. Se procede si se encuentra un template más del 25% de identidad con el target. La confiabilidad del modelo decrese cuando la identidad de secuencias es pequeña y si el target-template tiene menos del 50% de identidad, se requiere de un ajuste manual del alineamiento.

32
Alignment mode Se inicia colocando una secuencia para alinear, especificando cual es el target y cual es la proteina estructuralmente conocida.

34
Project mode Se submite el modelo optimizado manualmente al servidor; es un DeepView project file. Contiene las estructuras sobrepuestas del template y el alineamiento entre el target y los templates.

36
Modelando proteinas 0ligomérica

45
Deep-View-Swiss-PBDViewer
Programa que tiene funciones integradas para la visualizacion, análisis y manipulación de proteínas en su secuencia y estructura. Se trabaja con PDB o ExPDB files que se bajan directamente del DeepView server.

50
Banco de datos donde se anotan los modelos de estructuras comparados en tres dimensiones que ha sido generado por el SWISS MODEL. Contiene modelos 3D, de secuencias del Swiss-Prot y TrEMBL.

54
Introducimos la secuencia problema.
Partimos de una secuencia aminoacídica lineal, por ejemplo: >secuencia-problema MSKVPRNFRL LEELEKGEKG FGPESCSYGL ADSDDITMTK WNGTILGPPH SNHENRIYSL SIDCGPNYPD SPPKVTFISK INLPCVNPTT GEVQTDFHTL RDWKRAYTME TLLLDLRKEM ATPANKKLRQ PKEGETF Una vez que tenemos la secuencia que queremos analizar, debemos introducirla en el programa SWISS-MODEL a través de su servidor web, accesible en la red. En este servidor debemos introducir además de la secuencia, nuestra dirección de "mail", nuestro nombre, y un título para el proceso SWISS-MODEL (First Approach mode).
.")
55
2) Recepción en nuestro mail de un mensaje confirmatorio.
Si el proceso ha comenzado sin ningún problema recibiremos un mensaje por mail donde, a parte de otras informaciones, están recogidos la fecha, el título de la búsqueda y el código de identificación de nuestro proceso.

56
3) Recepción de un mail con los distintos procesos seguidos por SWISS-MODEL.
El segundo mensaje que recibimos desde el servidor de mail presenta una descripción abreviada de los procesos seguidos por el programa SWISS-MODEL. En realidad el programa SWISS-MODEL es el resultado de un conjunto de subprocesos, o programas más pequeños, los cuales son aplicados de forma secuencial. Diferenciaremos cada uno de los pasos seguidos por SWISS-MODEL utilizando como referente el mail de procesos.

57
- SWISS-MODEL realiza una búsqueda por homología de secuencia en una base de datos de secuencias de estructura conocidas, el programa utilizado es "BLASTP2" y la base de datos consultada "ExNRL-3D". Los resultados se muestran ordenados por la puntuación P(N) del BLAST.
 del BLAST..")
58
- Con el programa "SIM" Se seleccionan todos los "moldes" (estructuras 3D sobre las que nos basaremos para hacer nuestro modelo) con una identidad de secuencia superior al 25% y con una región alineada superior a 20 residuos. Adicionalmente, este programa detecta los posibles distintos dominios que debieran ser modelados a partir de moldes estructurales diferentes. Este proceso queda recogido del siguiente modo...

59
- En este paso se genera el fichero de entrada ("imput file") para el program "ProModII" el cual basándose en los moldes seleccionados (templates) de la base de datos "ExPDB" originará todos los modelos a partir de nuestra secuencia problema.
 para el program ProModII el cual basándose en los moldes seleccionados (templates) de la base de datos ExPDB originará todos los modelos a partir de nuestra secuencia problema.")
61
- Como último paso, mediante el programa "Gromos96", se optimiza/an el/los modelo/os basándose en un proceso de minimización de energía.

63
GRACIAS!

Presentaciones similares









































>")



