Descargar la presentación
La descarga está en progreso. Por favor, espere

1
HERENCIA NO TRADICIONAL
HERENCIA MITOCONDRIAL IMPRINTING GENÓMICO MUTACIONES DINÁMICAS: 1) EN REGIONES NO CODIFICANTES 2) POR POLIGLUTAMINAS MOSAICISMO 1) GONADAL 2) SOMÁTICO DISOMÍA UNIPARENTAL
 EN REGIONES NO CODIFICANTES. 2) POR. POLIGLUTAMINAS. MOSAICISMO 1) GONADAL. 2) SOMÁTICO. DISOMÍA UNIPARENTAL.")
2
Enfmermedades Mitocondriales

3
GENOMA MITOCONDRIAL 5 a10 moléculas de ADN idénticas 16.569 pb
37 genes Codifica: 2 ARNr 22 ARNt 13 polipépdidos La mayoría de las proteínas mitocondriales son codificadas por genes nucleares, sintetizadas en el citosol e importadas a la mitocondria

4
HERENCIA MITOCONDRIAL
Genoma extranuclear (citoplasmático) Teoría endosimbiótica Herencia materna Tasa de mutación 10 veces superior al genoma nuclear
 Teoría endosimbiótica. Herencia materna. Tasa de mutación 10 veces superior al genoma nuclear.")
5
PATOLOGÍA MITOCONDRIAL
Heteroplasmia u homoplasmia Encefalomiopatías (alta dependencia del metabolismo oxidativo) Envejecimiento? Mutaciones en el genoma nuclear pueden provocar patologías mitocondriales
 Envejecimiento Mutaciones en el genoma nuclear pueden provocar patologías mitocondriales.")
6
MERRF Myoclonic Epilepsy and Ragged-Red Fibers.
Es una enfermedad asociada con una mutación en el ADNmt que codifica un ARNt. Se caracteriza clínicamente por: mioclonía, ataxia, convulsiones generalizadas, debilidad muscular antes de los 20 años y anatomopatológicamente por Ragged-red fibers que son mitocondrias “cristalizadas” en el sarcolema).
.")
7
Imprinting Genómico

8
IMPRINTING Mecanismo normal de regulación de la expresión de algunos genes. Expresión monoalélica en un determinado locus, haciendo que la expresión dependa de que su origen sea paterno o materno. Fenómenos epigenéticos (reversibles, no mutacionales). Metilación y cambios en la estructura local de la cromatina de forma diferencial en ciertas regiones genómicas. Muchos genes que sufren imprinting están ubicados en clusters en determinadas regiones cromosómicas y regulados coordinadamente (dominios y centros de imprinting). Dos clusters principales en el genoma humano: Cluster ANGELMAN/PRADER-WILLI (15q11-13) Cluster H19/Igf2 (11p15)
. Metilación y cambios en la estructura local de la cromatina de forma diferencial en ciertas regiones genómicas. Muchos genes que sufren imprinting están ubicados en clusters en determinadas regiones cromosómicas y regulados coordinadamente (dominios y centros de imprinting). Dos clusters principales en el genoma humano: Cluster ANGELMAN/PRADER-WILLI (15q11-13) Cluster H19/Igf2 (11p15)")
9
IMPRINTING Cluster ANGELMAN/PRADER-WILLI

10
SÍNDROME DE PRADER - WILLI
Características clínicas: Hipotonía central. Problemas de la alimentación y/o falla de crecimiento durante la infancia. Ganancia rápida de peso entre los 6 meses y el año. Hiperfagia. Rasgos faciales característicos: Diámetro bitemporal angosto, hendiduras palpebrales almendradas, comisura bucal hacia abajo. Hipogonodismo: Hipoplasia genital: labios menores y Clítoris pequeño en las mujeres; escroto hipoplásico y criptorquidia en varones. Pubertad tardía e incompleta. Infertilidad. Retraso del desarrollo/RM leve

11
SÍNDROME DE ANGELMAN Epilepsia. Dificultades del aprendizaje.
Marcha inestable o atáxica. Estado de ánimo alegre.

12
Mecanismo patogénico:
Pérdida de la función de alelos regulados por imprinting

13
DIAGNÓSTICO EN SD. DE PRADER-WILLI

14
Mutaciones Dinámicas

15
ENFS. POR REPETICIÓN DE TRIPLETAS EN REGIONES NO CODIFICANTES:
MUTACIONES DINÁMICAS ENFS. POR REPETICIÓN DE TRIPLETAS EN REGIONES NO CODIFICANTES: SÍNDROME DE X FRAGIL DISTROFIA MIOTÓNICA ATAXIA DE FRIEDREICH ENF. POR POLIGLUTAMINAS: COREA DE HUNTINGTON

16
S. DE X-FRAGIL Estatura entre 50 y 97p Macrocefalia / Dolicocefalia
Facies alargada Frente alta, prominente Puente nasal y nariz anchos Mandíbula prominente Orejas grandes y antevertidas Paladar ojival / hendido Dientes superpuestos, irregulares

17
S. DE X-FRAGIL Fenotipo conductual de varones
Déficit atencional Hiperactividad Perseveración Dif. en articulación verbal Lenguaje incomprensible Episodios de leng. repetitivo Trast. del comportamiento Timidez extrema/aislamiento Estereotipias y tics Signos autistas Autismo

18
S. DE X- FRAGIL Cromosoma X La mutación responsable del S. De X-frágil es una expansión inestable de los repetidos CGG en la región 5’ no traducida del gen FMR -1 localizado en el brazo largo del cromosoma X.

19
FENÓMENO DE ANTICIPACIÓN
La expansión del número de tripletes repetidos en el locus responsable de la enfermedad es la causa del fenómeno de ANTICIPACIÓN que consiste en: aumento en la severidad de los síntomas y/o un aumento de la penetrancia edad de inicio más temprana

20
DISTROFIA MIOTÓNICA Inicio tercera década
Clásico: miotonía, cambios distróficos en tejidos musculares, defectos de la conducción cardíaca Congénito: hipotonía, retardo mental, parálisis facial bilateral, disfagia y malformaciones

21
DISTROFIA MIOTÓNICA (CTG)n 3’ UTR Gen DMPK (CTG)n >1000, fenotipo congénito 100< (CTG)n <1000, fenotipo clásico 50< (CTG)n <100, fenotipo leve Cromosoma 19 5’ 3’ La mutación responsable de la DM1 es una expansión inestable de los repetidos CTG en la región 3’ no traducida del gen DMPK localizado en la banda 13.3 del brazo largo del cromosoma 19. El número de repetidos es altamente polimórfico en la población (mutación dinámica).
n <100, fenotipo leve. Cromosoma 19. 5’ 3’ La mutación responsable de la DM1 es una expansión inestable de los repetidos CTG en la región 3’ no traducida del gen DMPK localizado en la banda 13.3 del brazo largo del cromosoma 19. El número de repetidos es altamente polimórfico en la población (mutación dinámica).")
22
ATAXIA DE FRIEDREICH La Enfermedad de Friedreich es una enfermedad neurodegenerativa de herencia autosómica recesiva. Los pacientes con Enfermedad de Friedreich que tienen mutaciones identificables en el gen X25/FRDA son considerados como portadores de Enfermedad de Friedreich tipo 1. Aproximadamente el 96% de estos pacientes son homocigotos para una expansión de la tripleta GAA en el intron 1 del gen. Aproximadamente el 4% de los pacientes con Enfermedad de Friedreich son heterocigotos para una expansión GAA en el rango patológico en un alelo y una mutación puntual en el otro alelo.

23
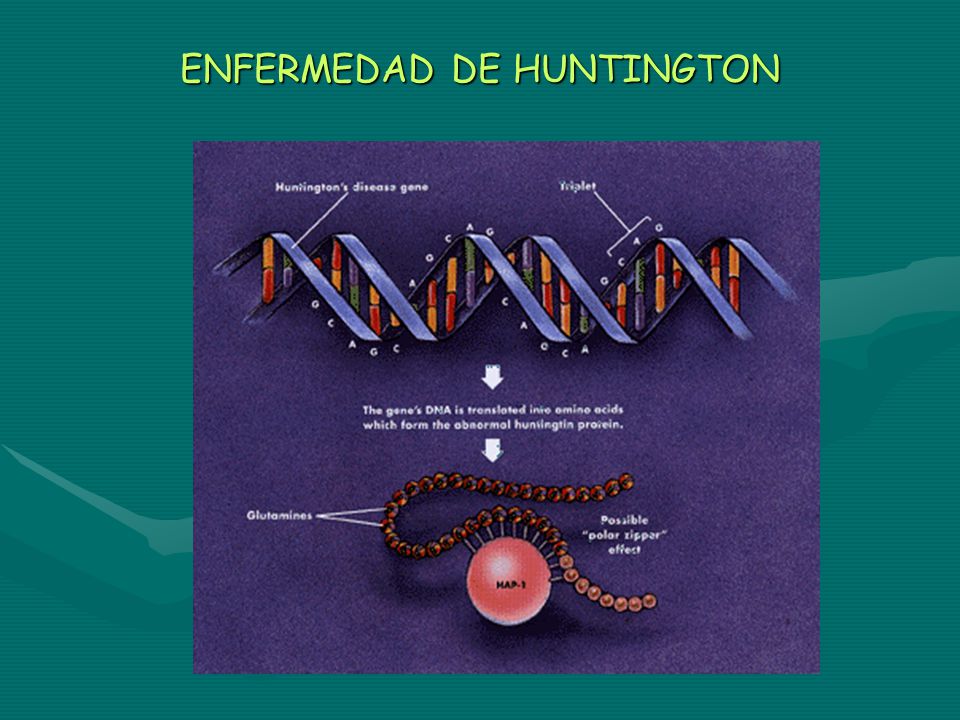
ENFEMEDAD DE HUNTINGTON
Defecto autosómico dominante, de penetrancia completa, pero cuya edad de comienzo varía entre los 20 y 50 años variando de forma lineal con el número de repeticiones heredadas en el Gen HD (4p16). Se asocia con una degeneración de neuronas GABAérgicas. Como la amplificación de tripletas al pasar de generación en generación es menor que en otros trastornos se comporta como un rasgo AD típico.
. Se asocia con una degeneración de neuronas GABAérgicas. Como la amplificación de tripletas al pasar de generación en generación es menor que en otros trastornos se comporta como un rasgo AD típico.")
24
ENFERMEDAD DE HUNTINGTON
25
Disomía Uniparental

26
DISOMÍA UNIPARENTAL Este tipo de herencia se da cuando ambos cromosomas de un par homólogo son heredados de uno de los padres como resultado de una no disyunción cromosómica. Por lo tanto es más probable que esto ocurra como resultado de la unión de un gameto con dos cromosomas de un par y uno normal, con posterior pérdida de un cromosoma extra en las primeras fases del desarrollo. Ejs.: Sd. de Silver Russell. Prader-Willi y Angelman. S. De Beckwith-Wiedemann. Fibrosis Quística (AR). Hemofilia A transmitida de padre a hijo.
. Hemofilia A transmitida de padre a hijo.")
27
Mosaicismo

28
MOSAICISMO Se define como la presencia en un individuo de dos líneas celulares, que difieren genéticamente pero que derivan de un mismo cigoto. SOMÁTICO Neurofibromatosis segmentaria Cáncer GONADAL Puede provocar que un progenitor cuyas células sanguíneas (somáticas) muestren un genotipo normal, tenga hijos afectados o portadores, según la mutación sea dominante o recesiva, e hijos sanos.
 muestren un genotipo normal, tenga hijos afectados o portadores, según la mutación sea dominante o recesiva, e hijos sanos.")
29
NUEROFIBROMATOSIS 1

30
MOSAICISMO GONADAL PC normal al nacimiento.
Periodo prenatal y perinatal aparentemente normal. PC normal al nacimiento. Desarrollo aparentemente normal durante los primeros 6 meses de vida. Desaceleración del crecimiento del PC en cualquier momento entre los 3 y 48 meses. Pérdida de las habilidades manuales adquiridas entre los 5 y 30 meses, desarrollo de movimientos estereotipados de las manos. Deterioro severo del lenguaje expresión y de comprensión y retardo psicomotor. Apraxia de la marcha y ataxia de tronco que aparece entre los 12 y 48 meses de vida.

Presentaciones similares



















