Descargar la presentación
La descarga está en progreso. Por favor, espere

5
pH=6 pH=2 10 2 4 6 8 12

7
Chairman: R. A. Marcus Speaker: Cyrus Levinthal Notes by: A. Rawitch
Mossbauer Spectroscopy in Biological Systems: Proceedings of a meeting held at Allerton House, Monticello, Illinois. Editors: J. T. P. DeBrunner and E. Munck University of Illinois Press (1969) Pages 22-24 How to Fold Graciously Chairman: R. A. Marcus Speaker: Cyrus Levinthal Notes by: A. Rawitch
 Pages How to Fold Graciously. Chairman: R. A. Marcus Speaker: Cyrus Levinthal Notes by: A. Rawitch.")
8
La paradoja de Levinthal
El número de conformaciones 3D diferentes que puede adoptar una cadena de 200 aminoácidos es:

9
Paradoja de Levinthal Si el tiempo mínimo para que ocurra una transición conformacional es de 1 picosegundo, entonces para explorar 1095 conformaciones habrá que esperar 1083 segundos (1075 años). Si la proteína tuviera que buscar al azar entre todas las conformaciones posibles hasta encontrar la nativa, el plegado no podría ocurrir en tiempos biológicos.
. Si la proteína tuviera que buscar al azar entre todas las conformaciones posibles hasta encontrar la nativa, el plegado no podría ocurrir en tiempos biológicos.")
10
Postulados El plegado no es una búsqueda al azar
Tienen que existir vías con intermediarios parcialmente plegados

11
Los modelos para el mecanismo de plegado proteico deben resolver la paradoja de Levinthal

12
Nucleation-condensation mechanism

18
27mero Interaccion fuerte (H-H): sin lineas Interacción débil (H-P, PP): linea de puntos A.1: no frustrada (28 interacciones fuertes) B.1: frustrada (debe resignarse a tener alguna interacción débil) Q=ctos/ctos ‘nativos’ Plegado eficiente: muchos estados de energía baja y Q alto Plegado ineficiente: muchos estados de energía baja y Q bajo
 B.1: frustrada. (debe resignarse a tener. alguna interacción débil) Q=ctos/ctos ‘nativos’ Plegado eficiente: muchos estados de. energía baja y Q alto. Plegado ineficiente: muchos estados de. energía baja y Q bajo.")
19
Folding is not a general property of heteropolymers
Folding is not a general property of heteropolymers. Heteropolymers, due to their many degrees of freedom and the many geometric constraints among them, are said to present a great frustration, that is, there is not a single conformation of the chain which optimizes all the interactions at the same time. In any conformation, the different interactions are conflicting, i.e., frustrated. In polypeptides, as in many heteropolymers, frustration is mainly due to chain connectivity between monomers

20
La “nueva visión” del plegado
Acepta que el número de conformaciones es astronómico Rechaza la existencia de vías de plegado Incorpora el concepto de flujo conformacional No es un modelo, es una metáfora que puede ayudar a pensar el problema del plegado

26
Estos modelos demostraron que puede encontrarse un mínimo energético sin realizar una ‘busqueda’ exhaustiva del espacio conformacional. Es decir, resolvieron la paradoja de Levinthal para un polímero ideal. Si esto se aplica a las proteínas todavía debe ser demostrado experimentalmente, pero hay consenso de que es relevante.

27
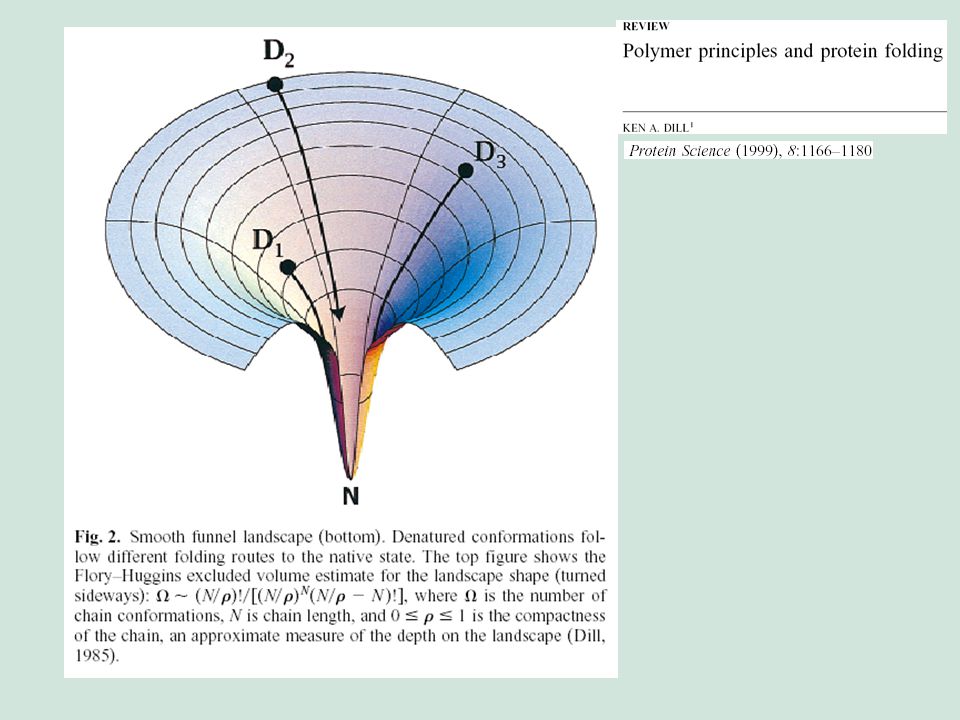
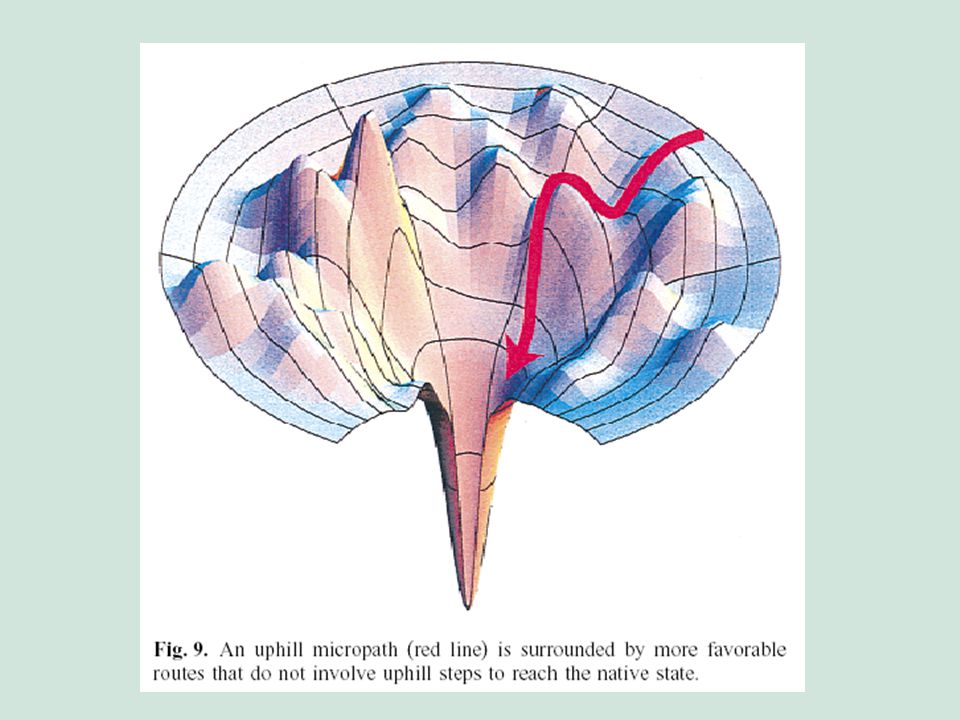
El paisaje conformacional
El principio de frustración El gap energético El flujo conformacional debido a una preferencia energética por el estado nativo

34
El problema del plegado
? El problema del plegamiento Existe una relación compleja entre la información conformacional de un polipéptido y su estructura tridimensional. Al conjunto de secuencias le corresponde, de alguna manera, un conjunto de estructuras para cada condición experimental. Los experimentos que vamos a ver abordan específicamente esa relación. conjunto de estructuras conjunto de secuencias

35
El número de cadenas diferentes de longitud l que se pueden construir con n tipos de aminoácidos es:
Una proteína típica tiene 200 residuos. El número posible de proteínas diferentes de 200 residuos es: 10260

36
El “Problema del Plegado Proteico” consiste en deducir la estructura 3D que adoptará una dada secuencia de aminoácidos

37
Premio Nobel de Química 1972
C. Anfinsen Premio Nobel de Química 1972 La secuencia de aminoácidos determina la estructura 3D

38
Las proteínas son una clase homogénea de objetos que almacena sistemáticamente información en su secuencia y en su forma La información en la secuencia depende de interacciones covalentes y determina la forma La forma depende de interacciones no covalentes está codificada en la secuencia y determina la función

39
Dependencia de la estructura 3D con la secuencia
j y y son los ángulos dihedros de cada enlace peptídico y x representa 1 de los 20 aminoácidos normalmente encontrados en las proteínas, indexados por la posición en la secuencia

40
lysozyme and in the other fatty acid binding protein
Seq 1: 131 residues Seq 2: 131 residues I am showing here two different structures belonging to two proteins of 131 residues. The information content of each sequence is what makes one to become lysozyme and the other fatty acid binding protein The information content of the sequence determines that in one case you get lysozyme and in the other fatty acid binding protein

41
“Not all the residues carry the same amount
of conformational information...” However, the conformational information present in the sequence is, at best, diffuse, and no all the residues carry the same amount of information. Alexander et al. (2005) Biochemistry 44, and references therein
 Biochemistry 44, and references therein.")
42
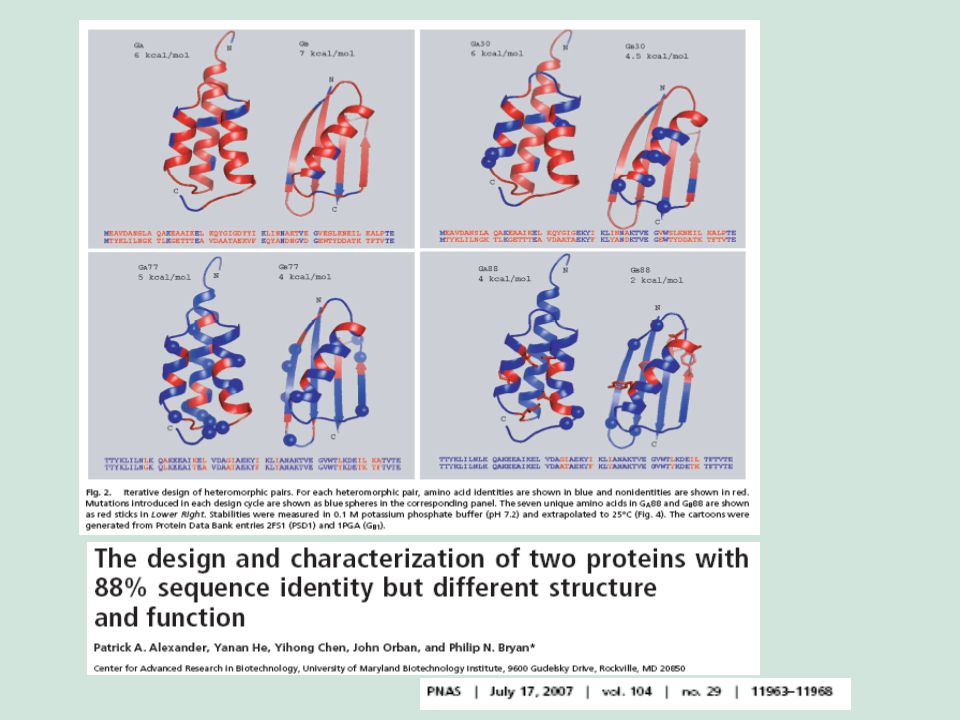
Alexander et al. (2005) Biochemistry 44, 14045-14054
Let me clarify this with an example from the literature. Wild-type Protein A and G have 14% of identical residues. By in vitro mutagenesis and selection these proteins were evolved to yield variants that have 60% identical residues and still different structure. Alexander et al. (2005) Biochemistry 44,
 Biochemistry 44,")
44
“Proteins tolerate a surprisingly large number of mutations...
There are several examples of proteins with no sequence homology belonging to the same structural class... Identical sequences of 8–11 residues adopt different structures in different proteins...” Other experimental results indicate that: (a) Proteins are remarkable resistant to mutations. (b) There are several examples of proteins with no sequence similarity that belong to the same structural class. (c) Chameleon sequences of 8-11 residues adopts different structure in different proteins Gebhard et al. (2006) J. Mol. Biol. 358, and references therein
 Proteins are remarkable resistant to mutations. (b) There are several examples of proteins with no sequence similarity that belong to the same structural class. (c) Chameleon sequences of 8-11 residues adopts different structure in different proteins. Gebhard et al. (2006) J. Mol. Biol. 358, and references therein.")
45
Los módulos se pueden eliminar alternativamente sin impedir el plegado
Las proteínas parecen estar formadas por módulos de plegado independientes. Cada uno de estos módulos tiene la potencialidad de plegarse en un número limitado de formas. No hay intercambio esencial de información entre módulos (ninguno determina la conformación de otro módulo). La estructura que adoptará cada módulo depende de la estabilidad del conjunto de modulos cuando se encuentran plegados. Los módulos se pueden eliminar alternativamente sin impedir el plegado
. La estructura que adoptará cada módulo depende de la estabilidad del conjunto de modulos cuando se encuentran plegados. Los módulos se pueden eliminar alternativamente sin impedir el plegado.")
46
edad del universo = 1010 años tiempo estimado para ‘visitar’ todas las conformaciones posibles de una proteína de 200 resiuos = 1083 años masa de la tierra = 1027 g = 1046 moléculas proteicas de 200 residuos masa del universo = 1060 g = 1079 moléculas proteicas de 200 residuos Una muestra que contenga todas las secuencias posibles de 200 residuos (10260) tendría una masa de universos Las secuencias naturales (existentes) = 108 El número de formas proteicas básicas conocidas = 104 La probabilidad de generar al azar una forma proteica básica =
 tendría una masa de universos. Las secuencias naturales (existentes) = 108. El número de formas proteicas básicas conocidas = 104. La probabilidad de generar al azar una forma proteica básica =")
47
Las proteínas naturales son un subconjunto de las proteínas posibles que cumple con lo siguiente:
Mínima frustración Colapsan rapidamente a un conjunto grande de conformaciones que son competentes para llegar al estado nativo en forma rápida La evolución habría seleccionado esas secuencias No sabemos qué propiedad de la secuencia es la que confiere las características descriptas

Presentaciones similares
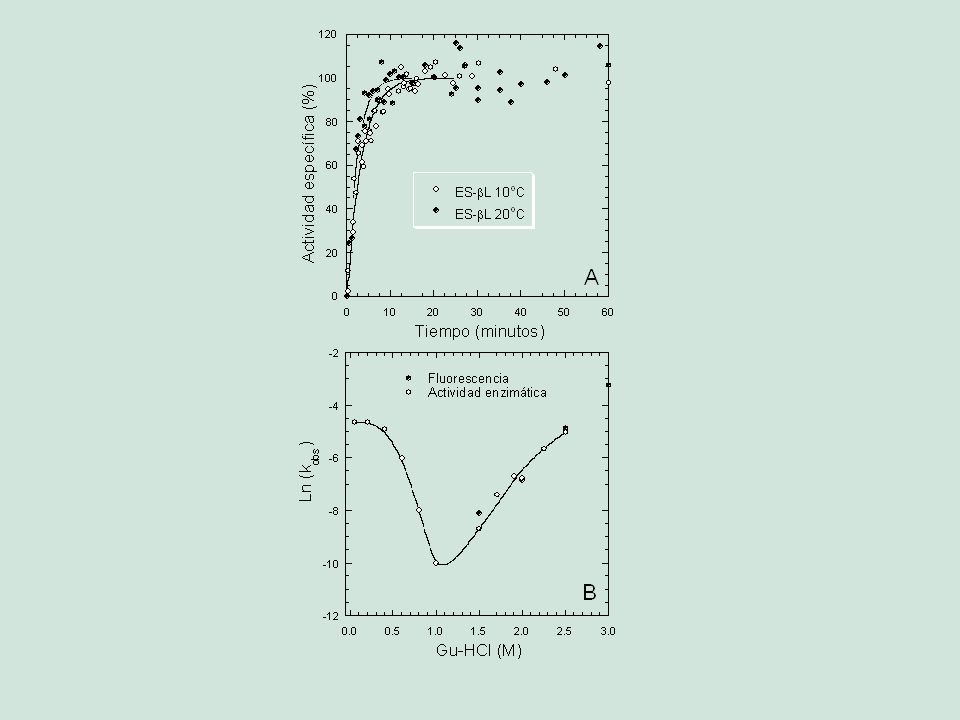
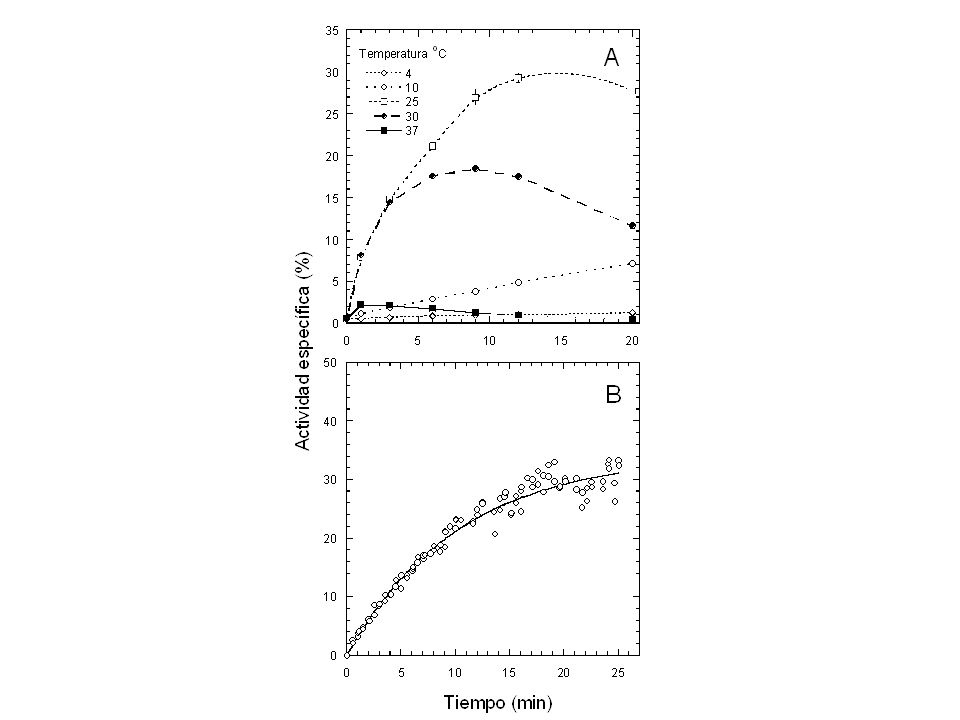























 Compactación óptima Grupos polares en el exterior Mínima superficie expuesta Anisotropía y baja periodicidad Organización jerárquica.>")




