Descargar la presentación
La descarga está en progreso. Por favor, espere

1
Tema 1-Introducción y Perspectivas
QUIMICA ORGÁNICA Tema 1-Introducción y Perspectivas La Química Orgánica es la parte de la Química que estudia los Compuestos del Carbono Importancia Más del 95% de las sustancias químicas conocidas son compuestos del carbono. Todos los compuestos responsables de la vida (ácidos nucleicos, proteínas, enzimas, hormonas, azúcares, lípidos, vitaminas, etc.) son sustancias orgánicas. El progreso de la Química Orgánica permite profundizar en el esclarecimiento de los procesos vitales La industria química (fármacos, polímeros, pesticidas, herbicidas, etc.) juega un papel muy importante en la economía mundial e incide en muchos aspectos de nuestra vida diaria con sus productos.
 son sustancias orgánicas. El progreso de la Química Orgánica permite. profundizar en el esclarecimiento de los procesos vitales. La industria química (fármacos, polímeros, pesticidas, herbicidas, etc.) juega un papel muy importante en la economía mundial. e incide en muchos aspectos de nuestra vida diaria con sus productos.")
2
El estudio de los compuestos de carbono comprende varias facetas :
QUIMICA ORGÁNICA El estudio de los compuestos de carbono comprende varias facetas : ESTRUCTURA MOLECULAR elucidación estructural REACTIVIDAD QUÍMICA Mecanismos de reacción SÍNTESIS Diseño de métodos eficientes RELACIONES CON OTRAS ÁREAS FARMACIA MEDICINA BIOLOGÍA APLICACIONES Desarrollo industrial, biológico, médico QUIMICA ORGÁNICA

3
Conformación bioactiva estructura-actividad Forma de administración
QUIMICA ORGÁNICA REACTIVIDAD QUÍMICA PROPIEDADES FÍSICAS ESTRUCTURA MOLECULAR ACTIVIDAD BIOLÓGICA SÍNTESIS DE FÁRMACOS DISEÑO DE FÁRMACOS Conformación bioactiva Correlaciones estructura-actividad Forma de administración Metabolismo

4
QUIMICA ORGÁNICA Ácido o-acetoxibenzoico Ácido acetilsalicílico
N-(4-hidroxifenil)acetamida Ácido o-acetoxibenzoico Ácido acetilsalicílico Aspirina Paracetamol ESTRUCTURA MOLECULAR
acetamida. Ácido o-acetoxibenzoico. Ácido acetilsalicílico. Aspirina. Paracetamol. ESTRUCTURA. MOLECULAR.")
5
QUIMICA ORGÁNICA FUENTES AGENTES DE DIAGNOSIS INMUNOTERAPIA
AGENTES TERAPÉUTICOS FUENTES 1. PURIFICACIÓN DE PRINCIPIOS ACTIVOS ADORMIDERA OPIO 2. MODIFICACIÓN DE LOS PRODUCTOS NATURALES 3. PREPARACIÓN SINTÉTICA Seturner (1817) Pelletier Caventou (1817) Crum-Brown Fraser (1869) F. Hoffman (1898) Morfina
 Pelletier. Caventou (1817) Crum-Brown. Fraser (1869) F. Hoffman. (1898) Morfina.")
6
QUIMICA ORGÁNICA ESTRUCTURA MOLECULAR

7
QUIMICA ORGÁNICA ESTRUCTURA MOLECULAR

8
QUIMICA ORGÁNICA ESTRUCTURA MOLECULAR MÉTODOS TEÓRICOS
MÉTODOS MECANO-CUÁNTICOS MÉTODOS DE CAMPOS DE FUERZAS

9
Tema 2-Estructura Electrónica de las
QUIMICA ORGÁNICA Tema 2-Estructura Electrónica de las Moléculas Orgánicas

10
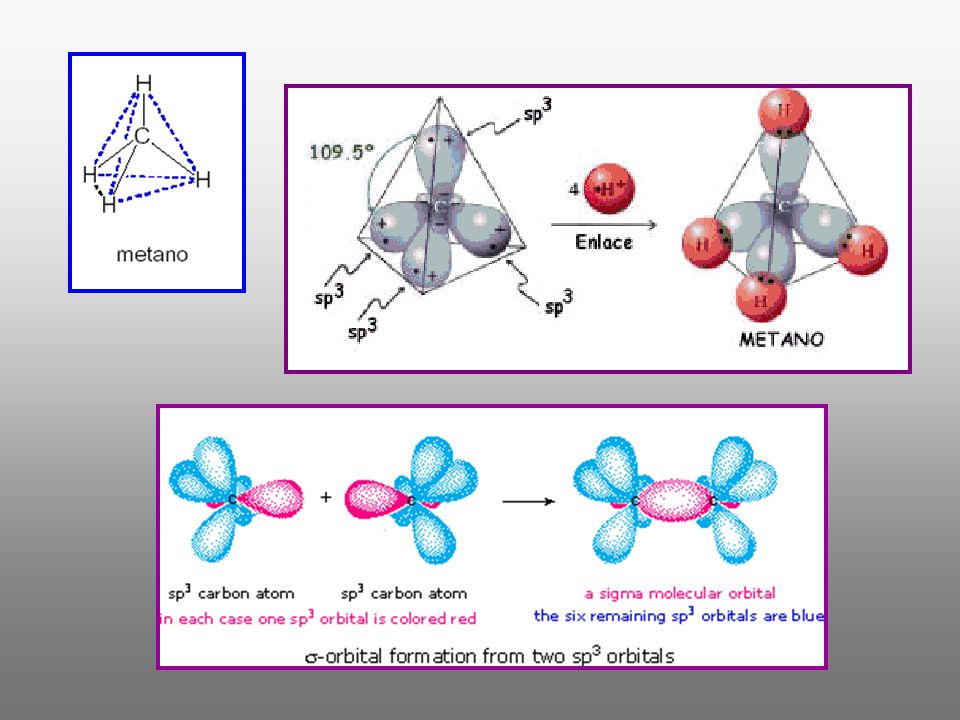
TEORÍA ESTRUCTURAL Hace referencia a una serie de conceptos que nos explican como se unen los átomos para dar lugar a las moléculas ¿Que fuerzas los mantienen unidos?.-Concepto: los enlaces químicos. ¿Cómo están distribuidos los electrones?.-Concepto: los orbitales, que pueden ser atómicos (la distribución de electrones en un átomo) o moleculares (la distribución de electrones en una molécula). ¿A que distancia y a qué ángulo se encuentra un átomo con respecto a otros en una molécula? Concepto: la estructura tridimensional de la molécula, que da lugar a la rama de la estereoquímica. ¿De que manera podemos representarlos?.-Concepto: las diversas formas de representar los electrones, enlaces y moléculas en el papel; modelos moleculares.
 o moleculares (la distribución de electrones en una molécula). ¿A que distancia y a qué ángulo se encuentra un átomo con respecto a otros en una molécula Concepto: la estructura tridimensional de la molécula, que da lugar a la rama de la estereoquímica. ¿De que manera podemos representarlos .-Concepto: las diversas formas de representar los electrones, enlaces y moléculas en el papel; modelos moleculares.")
11
ESTRUCTURAS DE LEWIS El símbolo representa el núcleo y los electrones internos Los electrones de valencia se representan por puntos (coincide con el grupo del Sistema Periódico) Hidrógeno Oxígeno Cloro (6e- de valencia) (7e- de valencia) Los electrones de valencia se distribuyen alrededor del átomo central Electrones enlazantes (compartidos) y no enlazantes (electrones no compartidos)
 Hidrógeno. Oxígeno. Cloro. (6e- de valencia) (7e- de valencia) Los electrones de valencia se distribuyen alrededor del átomo central. Electrones enlazantes (compartidos) y no enlazantes (electrones no compartidos)")
12
- NH2 CH4 H3O + 1) Dibujar correctamente el esqueleto molecular
indicando los átomos conectados por enlaces (Los H siempre son periféricos) 2) Contar el nº de e- de valencia disponibles Sumar 1 e- por cada carga negativa (anión) Restar 1 e- por cada carga positiva (catión) H3O + - NH2 CH4
 2) Contar el nº de e- de valencia disponibles. Sumar 1 e- por cada carga negativa (anión) Restar 1 e- por cada carga positiva (catión) H3O + - NH2. CH4.")
13
3) Dibujar enlaces covalentes entre todos los átomos, de forma que el mayor nº de átomos posean octetes (excepto el H, doblete)
 Dibujar enlaces covalentes entre todos los átomos, de forma que el mayor nº de átomos posean octetes (excepto el H, doblete)")
14
N2 O2 BF3 Para completar octetes puede ser necesario compartir
dos o más pares electrones N2 O2 BF3 A veces la regla del octete no se cumple Si el número de electrones no es suficiente para completar todos los octetes se completan primero los de los elementos más electronegativos

15
Carga formal Cálculo de las cargas formales de los átomos en una estructura de Lewis: En el caso de iones, la suma de las cargas formales de los diferentes átomos coincide con la carga del ion. CF = nº electrones de valencia - nº electrones no enlazantes - 1/2 (electrones enlazantes) Carga Formal de H = = 0 Carga Formal de O (enlace sencillo) = 6 - (2 + 4) = 0 Carga Formal de O (enlace doble) = 6 - (2 + 4) = 0 Carga Formal de N = 5 - (3 + 2) = 0 Carga Formal de H = = 0 Carga Formal de N = = +1 Carga Formal de O (enlace doble) = 6 - (2 + 4) = 0 Carga Formal de O (enlace sencillo) = 6 - (1+ 6)= -1 Carga Formal de C =4 - 4 = 0 -En algunos casos las reglas anteriores dan lugar a estructuras de Lewis con separación de cargas O- NR3 + -
 Carga Formal de H = = 0 Carga Formal de O (enlace sencillo) = 6 - (2 + 4) = 0 Carga Formal de O (enlace doble) = 6 - (2 + 4) = 0 Carga Formal de N = 5 - (3 + 2) = 0. Carga Formal de H = = 0 Carga Formal de N = = +1 Carga Formal de O (enlace doble) = 6 - (2 + 4) = 0 Carga Formal de O (enlace sencillo) = 6 - (1+ 6)= -1 Carga Formal de C =4 - 4 = 0. -En algunos casos las reglas anteriores. dan lugar a estructuras de Lewis con. separación de cargas. O- NR")
17
INTERPRETACIÓN DEL ENLACE COVALENTE MEDIANTE
LA TEORÍA DE ORBITALES MOLECULARES C 1s 2 2s 2p Configuración electrónica Estado fundamental

18
TEORÍA DE ORBITALES MOLECULARES
MÉTODO CLOA El solapamiento entre OAs es más efectivo cuanto menor sea la diferencia de energía entre ambos. La E de estabilización es menor que la de desestabilización Al solaparse los orbitales se puede sumar o restar la densidad electrónica Cuando se suman se aumenta la densidad electrónica entre los núcleos, dando lugar a una relación enlazante y a un orbital molecular de menor energía que los orbitales atómicos. Por el contrario, cuando se restan, disminuye la densidad electrónica entre los núcleos. Esto conduce a una condición inestable en donde los núcleos se repelen y el orbital resultante tiene mayor energía que cualquiera de los orbitales atómicos que lo componen.

19
Interacciones s-p: enlaces s
Solapamiento fuera de fase Solapamiento en fase Interacciones s-p: enlaces s

20
Interacciones s-p: enlaces s
Solapamiento nulo Solapamiento frontal s-p

21
Interacciones p-p Solapamiento frontal sp-p

22
Interacciones p-p pp-p Solapamiento lateral

24
TEORÍA DEL ENLACE DE VALENCIA
Enlace s Enlace p

25
TIPOS DE HIBRIDACIÓN EN
COMPUESTOS ORGÁNICOS

31
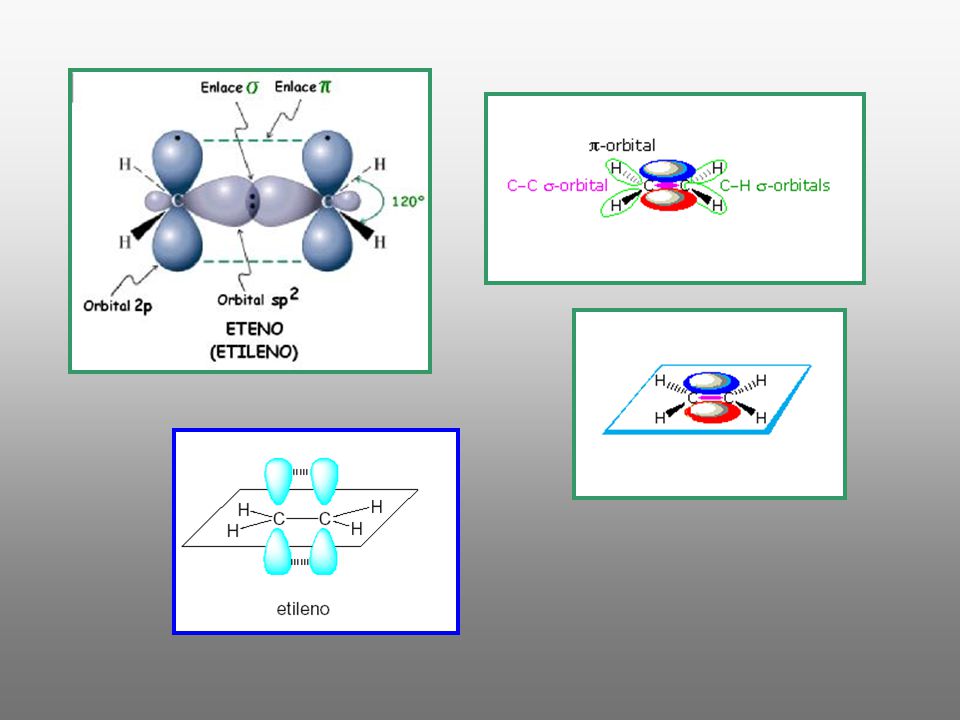
Electronegatividad del carbono en función de su hibridación
El carbono tiene mayor electronegatividad a medida que aumenta el carácter s de la hibridación. Por tanto los carbonos del etano (sp3) son menos electronegativos que los del eteno (sp2) y éstos a su vez menos electronegativos que los del etino (sp). Al pasar del etano al etino disminuye la densidad electrónica de los átomos de hidrógeno y por tanto aumenta su acidez
 son menos electronegativos que los del eteno (sp2) y éstos a su vez menos electronegativos que los del etino (sp). Al pasar del etano al etino disminuye la densidad electrónica de los átomos de hidrógeno y por tanto aumenta su acidez.")
32
Sistemas conjugados: estructura, estabilidad y reactividad
HOMO LUMO

33
Tema 3-Clasificación y Nomenclatura de los Compuestos Orgánicos
QUIMICA ORGANICA Tema 3-Clasificación y Nomenclatura de los Compuestos Orgánicos BIBLIOGRAFÍA Formulacion y Nomenclatura. E. Quiñoá, y R. Riguera, Nomenclatura y Representación de los Compuestos Orgánicos. Una guia de estudio y autoevaluación, McGraw-Hill, 1996. W.R. Peterson, Formulación y nomenclatura en Química Orgánica, 15ª Ed., Edunsa, 1993. Historicamente arbitraria (ej, ác. láctico, fórmico etc..) nombres vulgares Sistematización IUPAC (Unión Internacional de Química Pura y Aplicada)
 nombres vulgares. Sistematización IUPAC (Unión Internacional de Química Pura y Aplicada)")
34
Concepto de Radical, Grupo Funcional
y Serie homóloga

35
Concepto de Radical, Grupo Funcional
y Serie Homóloga Radical: Agrupamiento atómico con una valencia libre Grupo funcional: Atomo o grupo de átomos responsables de la reactividad química y propiedades Serie Homóloga: Conjunto de compuestos con el mismo grupo funcional y diferente tamaño del radical hidrocarbonado

36
Principales Series Homólogas

37
Compuestos Orgánicos Serie acíclica Serie cíclica Alicíclica Aromática Carbocíclica Heterocíclica Alicíclica Aromática

38
¿Cómo representar las estructuras?

39
NO PERMITEN DISTINGUIR ENTRE ISÓMEROS:
FÓRMULAS MOLECULARES, constituidas por SÍMBOLOS ATÓMICOS: Indican los elementos que forman el compuesto SUBÍNDICES: Indican el número de cada tipo de átomos en la molécula Ejemplos: C6H8, C4H9BrO, C6H12, C6H12O6, ... No informan sobre: Conectividad entre átomos (enlaces) Distribución espacial NO PERMITEN DISTINGUIR ENTRE ISÓMEROS: Compuestos que, con la misma fórmula molecular , tienen distinta estructura RECURRIR A REPRESENTACIONES MÁS PRECISAS
 Distribución espacial. NO PERMITEN DISTINGUIR ENTRE ISÓMEROS: Compuestos que, con la misma fórmula molecular , tienen distinta estructura. RECURRIR A REPRESENTACIONES MÁS PRECISAS.")
40
ISÓMEROS ESTRUCTURALES ESTEREOISÓMEROS
ESQUELETO POSICIÓN FUNCIÓN ISÓMEROS ESTRUCTURALES ESTEREOISÓMEROS CONFORMACIONALES CONFIGURACIONALES: ENANTIÓMEROS DIASTEREÓMEROS Al aumentar el nº de C aumenta en nº de isómeros

41
ISÓMEROS ESTRUCTURALES (CONSTITUCIONALES)
Tienen diferente conectividad entre los átomos que forman la molécula ISÓMEROS DE ESQUELETO: Son el resultado de las distintas secuencias de los átomos de carbono permisibles según la Teoría Estructural Ejemplos: C5H12 C5H10

42
ISÓMEROS DE POSICIÓN: Son el resultado de colocar un mismo grupo funcional
en posiciones estructuralmente no equivalentes de un mismo esqueleto carbonado Ejemplo: C5H12O Ejemplos: ISÓMEROS DE FUNCIÓN: Son el resultado de las diversas ordenaciones atómicas permisibles que pueden dar lugar a grupos funcionales diferentes C3H6O C2H6O C2H4O2

43
Csp3 ESTEREOISÓMEROS: Isómeros con la misma conectividad de enlaces
que difieren en la disposición espacial de los átomos Csp3 EN EL PLANO DEBAJO DEL PLANO MODELOS MOLECULARES ENCIMA DEL PLANO

45
ESTEREOISÓMEROS ISÓMEROS CONFORMACIONALES
Interconvertibles por giro alrededor de enlaces sencillos

46
H axial BOTE H ecuatorial SILLA ISÓMEROS CONFORMACIONALES
Interconvertibles por giro alrededor de enlaces sencillos H ecuatorial H axial SILLA BOTE

48
ENANTIÓMEROS ISÓMEROS CONFIGURACIONALES
No Interconvertibles, para pasar de uno a otro se necesita un gran aporte de energía que suele suponer rotura y formación de enlaces covalentes ENANTIÓMEROS Estereoisómeros que son imágenes especulares no superponibles

49
ENANTIÓMEROS Estereoisómeros que son imágenes especulares no superponibles

50
DIASTEREÓMEROS O DIASTEREOISÓMEROS
Estereoisómeros que NO son imágenes especulares

51
Z (cis) E (trans) DIASTEREÓMEROS O DIASTEREOISÓMEROS
Estereoisómeros que NO son imágenes especulares Z (cis) E (trans)
 E (trans)")
52
C6H8 C4H9BrO FÓRMULAS DESARROLLADAS (Expandidas): Se representan todos
los átomos por sus símbolos y los enlaces que los unen por trazos C6H8 C4H9BrO

53
C6H8 FÓRMULAS SEMIDESARROLLADAS (Semiexpandidas o Semicondensadas):
Se omiten los enlaces con los hidrógenos y se indica el número de estos con un subíndice. A veces también se omiten los enlaces sencillos C-C C6H8

54
C4H9BrO FÓRMULAS SEMIDESARROLLADAS (Semiexpandidas o Semicondensadas):
Se omiten los enlaces con los hidrógenos y se indica el número de estos con un subíndice. A veces también se omiten los enlaces sencillos C-C C4H9BrO

55
C6H8 FÓRMULAS SIMPLIFICADAS: Se representan las cadenas carbonadas
mediante líneas en zig-zag en las que cada segmento representa un enlace y cada punto de unión un átomo de carbono Se omiten los átomos de hidrógeno unidos a carbono, pero sí se incluyen los heteroátomos y sus hidrógenos Los dobles y triples enlaces se representan con dos y tres segmentos C6H8

56
FÓRMULAS SIMPLIFICADAS: Se representan las cadenas carbonadas mediante líneas en zig-zag en las que cada segmento representa un enlace y cada punto de unión un átomo de carbono. Se omiten los átomos de hidrógeno unidos a carbono, pero sí se incluyen los heteroátomos y sus hidrógenos. Los dobles y triples enlaces se representan con dos y tres segmentos C4H9BrO

57
FÓRMULAS EN PERSPECTIVA
Las estructuras de los compuestos químicos son tridimensionales (Csp3 tetraédrico) Csp3 EN EL PLANO DEBAJO DEL PLANO ENCIMA DEL PLANO En las representaciones en perspectiva se utilizan: TRAZOS CONTINUOS Y GRUESOS para indicar los enlaces que se proyectan fuera del plano y hacia delante (por encima del plano del dibujo) TRAZOS DISCONTÍNUOS Y DE GROSOR NORMAL para enlaces que están dirigidos hacia atrás (por debajo del plano del dibujo) TRAZOS CONTINUOS DE GROSOR NORMAL para todo lo que está contenido en el plano del papel
 Csp3. EN EL PLANO. DEBAJO DEL PLANO. ENCIMA DEL PLANO. En las representaciones en perspectiva se utilizan: TRAZOS CONTINUOS Y GRUESOS para indicar los enlaces que se proyectan. fuera del plano y hacia delante (por encima del plano del dibujo) TRAZOS DISCONTÍNUOS Y DE GROSOR NORMAL para enlaces que están. dirigidos hacia atrás (por debajo del plano del dibujo) TRAZOS CONTINUOS DE GROSOR NORMAL para todo lo que está contenido. en el plano del papel.")
58
REPRESENTACIONES MIXTAS
Se recurre a ellas muchas veces por conveniencia o simplicidad cuando interesa resaltar la estructura de algún punto de la molécula Enantiómeros Diastereómeros Diastereómeros

59
CICLOHEXANO C6H12 sp3 C

60
BENCENO C6H6 sp2 C

62
HIDROCARBUROS: CLASIFICACIÓN Y NOMENCLATURA

63
Hidrocarburos: clasificación
Alcanos Alquenos Alquinos Alifáticos Acíclicos Alicíclicos o Cíclicos Cicloalcanos Cicloalquenos Cicloalquinos

64
Aromáticos Policíclicos Monocíclicos Aislados Condensados

65
Hidrocarburos Alifáticos Acíclicos:
Alcanos

66
ALCANOS NO RAMIFICADOS
Nombre: Prefijo que indica el nº de carbonos + ano Hepta + ano Heptano Me Et Pr Bu Abreviatura 1 Metano 2 Etano 3 Propano 4 Butano 5 Pentano 6 Hexano 7 Heptano 8 Octano 9 Nonano 10 Decano 11 Undecano 12 Dodecano 13 Tridecano 14 Tetradecano 15 Pentadecano 16 Hexadecano 17 Heptadecano 18 Octadecano 19 Nonadecano 20 Icosano 21 Henicosano 22 Docosano 23 Tricosano 24 Tetracosano 30 Triacontano 31 Hentriacontano 40 Tetracontano 50 Pentacontano 100 Hectano 115 Pentadecahectano

67
Radicales univalentes Nombre como sustituyente
Construcción del nombre -ANO ILO IL Numeración: Se comienza a numerar por el carbono que presenta la valencia libre Alcano de igual número de átomos de carbono Nombre como sustituyente Nombre del radical BUTANO BUTILO BUTIL

68
Nombres propios de alcanos ramificados y sus radicales
Radicales ramificados (Pri, i-Pr) (Bus, s-Bu) (Bui, i-Bu) (But, t-Bu) (i-Am) (t-Am) (Isoamilo) (Terc-amilo)
 (Bus, s-Bu) (Bui, i-Bu) (But, t-Bu) (i-Am) (t-Am) (Isoamilo) (Terc-amilo)")
69
Hidrocarburos acíclicos saturados ramificados: Estructuras complejas
I.- ELECCIÓN DE LA CADENA PRINCIPAL Hidrocarburos acíclicos saturados ramificados 1. La de mayor longitud (mayor nº átomos de C) 2. En caso de opción, la que posea: 2.1. Mayor número de cadenas laterales 2.2. Cadenas laterales con localizados más bajos 2.3. Mayor nº de C en cadenas laterales más cortas 2.4. Cadenas laterales menos ramificadas
 2. En caso de opción, la que posea: 2.1. Mayor número de cadenas laterales Cadenas laterales con localizados más bajos Mayor nº de C en cadenas laterales más cortas Cadenas laterales menos ramificadas.")
70
1. Elección de la cadena principal
NOMENCLATURA 1.1. Se elige la cadena de mayor número de átomos de carbono 1. Elección de la cadena principal 1* 8* 1 6 1 7 *Cadena principal

71
1.2. Aquella de mayor número de cadenas laterales
1 8 *3 cadenas laterales: CADENA PRINCIPAL 8* 1*

72
1.3. Aquella de cadenas laterales con localizador más bajo
8 carbonos 3 ramificaciones en 3, 4 y 6 1 3 6 8 4 *8 carbonos 3 ramificaciones en 2, 4 y 6 CADENA PRINCIPAL 1* 2*

73
1.4. Aquella de más carbonos en la cadena lateral más pequeña
9 carbonos 6 cadenas laterales posiciones 3,4 5,5,6,7 C en cadenas laterales 1,1,1,2,4,6 CADENA PRINCIPAL 1* 2* 3* 4* 9 carbonos 6 cadenas laterales posiciones 3,4 5,5,6,7 C en cadenas laterales 1,1,1,1,4,7 1 2 3 4 5 6 7 8 9 1.5. Aquella de cadenas laterales menos ramificadas

74
Numeración incorrecta
2. La numeración 2.1. Números más bajos a los sustituyentes 2, 3, 5 *NUMERACIÓN CORRECTA 2* 1* 3* 5* 2, 4, 5 Numeración incorrecta 1 2 4 5

75
Numeración incorrecta *E antes que M NUMERACIÓN CORRECTA
2.2. Números más bajos a los sustituyentes por orden alfabético 4-metil-7-etil 7 4 1 Numeración incorrecta 4-etil-7-metil *E antes que M NUMERACIÓN CORRECTA 4* 1* 7*

76
6-Etil-3-metil-5-propilnonano
3. El nombre Localizadores-Sustituyentes + Nombre Alcano (cadenas laterales) (cadena principal) 3.1. Se anteponen los nombres de los sustituyentes por orden alfabético acompañados de su localizador 6-Etil-3-metil-5-propilnonano 1 3 5 6 9
 (cadena principal) 3.1. Se anteponen los nombres de los sustituyentes por orden alfabético acompañados de su localizador. 6-Etil-3-metil-5-propilnonano")
77
3. 2. Sustituyentes repetidos en el mismo y/u otro carbono
3.2. Sustituyentes repetidos en el mismo y/u otro carbono repiten el número y utilizan prefijos multiplicativos (di-, tri-, tetra, etc) 2, 2, 4-Trimetilpentano 2 1 4 5
 2, 2, 4-Trimetilpentano")
78
3.4. Los prefijos n-, sec-, terc- no se alfabetizan
3.3. Los prefijos multiplicativos (di-, tri-, tetra, etc) no se alfabetizan 3.4. Los prefijos n-, sec-, terc- no se alfabetizan 3.5. Los prefijos iso, neo y ciclo si se alfabetizan y se escriben sin guión 4-terc-Butil-5-isobutil-2,7-dimetilnonano 4 1 2 5 7 9
 no se alfabetizan Los prefijos n-, sec-, terc- no se alfabetizan Los prefijos iso, neo y ciclo si se alfabetizan y se escriben sin guión. 4-terc-Butil-5-isobutil-2,7-dimetilnonano")
79
3. 6. Para nombrar a los radicales ramificados se aplican
3.6. Para nombrar a los radicales ramificados se aplican las mismas reglas. El C1 es el unido a la cadena principal. Los localizadores se escriben con primas o junto con el nombre del sustituyente entre paréntesis. Se alfabetiza el prefijo multiplicador del radical sencillo. Cuando hay varios radicales complejos se utiliza: bis, tris, tetrakis, etc, sin alfabetizar. 5,5-Bis(1,2-dimetilpropil)-6-(1,2,3-trimetilbutil)undecano 1 5 6 11 1’ 2’ 3’ 5,5-Bis-1’,2’-dimetilpropil-6-1’,2’,3’-trimetilbutilundecano
-6-(1,2,3-trimetilbutil)undecano ’ 2’ 3’ 5,5-Bis-1’,2’-dimetilpropil-6-1’,2’,3’-trimetilbutilundecano.")
80
Alquenos y Alquinos

81
1. Elección de la cadena principal
1.1. Aquella con mayor número de enlaces múltiples 1 enlace múltiple 1 9 *2 enlaces múltiples CADENA PRINCIPAL 1* 7*

82
1.2. Aquella de mayor longitud
*2 enlaces múltiples 8 carbonos CADENA PRINCIPAL 1* 8* 2 enlaces múltiples 7 carbonos 1 7

83
*2 enlaces múltiples 8 carbonos 2 dobles CADENA PRINCIPAL
1.3. Aquella con mayor número de enlaces dobles 2 enlaces múltiples 8 carbonos 1 doble y 1 triple 1 8 *2 enlaces múltiples 8 carbonos 2 dobles CADENA PRINCIPAL 1* 8*

84
2. Numeración 2.1. Números más bajos a los enlaces múltiples. En caso de igualdad los enlaces dobles tienen preferencia. 1 6 1* 6* *NUMERACIÓN CORRECTA

85
2.2. Números más bajos a los sustituyentes
1 6 8 1* 4* 8* *NUMERACIÓN CORRECTA

86
*E antes que M NUMERACIÓN CORRECTA
2.3. Números más bajos a los sustituyentes por orden alfabético 1 4 6 8 1* 4* 6* 8* *E antes que M NUMERACIÓN CORRECTA

87
3. El nombre Localizadores-Sustituyentes-Raiz Alcano (nº C cadena principal) -Localizadores-eno/ino 3.1. Se cambia -ano del alcano de igual número de átomos de carbono por -eno (alqueno) o por -ino (alquino) precedidos de un localizador que indica su posición 1* 3* 4* 5* 6* 3-Etil-4-metilhex-1-en-5-ino
 o por -ino (alquino) precedidos de un localizador que indica su posición. 1* 3* 4* 5* 6* 3-Etil-4-metilhex-1-en-5-ino.")
88
3. 2. Varios enlaces múltiples se indican con localizadores
3.2. Varios enlaces múltiples se indican con localizadores La terminación -eno se sustituye por -adieno, -atrieno, etc y -ino por -adiino,atriino, etc. 1 3 Buta-1,3-dieno

89
3.3. Dobles y triples enlaces: se indica el sufijo -eno antes que -ino
1 5 11 13 8 Trideca-1,8-dien-5,11-diino

90
3. 4. Isomería E-Z: Se indica la letra que indica la
3.4. Isomería E-Z: Se indica la letra que indica la configuración (E/Z) delante del nombre. Si hubiese varios, se escriben entre paréntesis y se indica su posición con localizadores. En caso de igualdad Z precede a E. 2 6 1 4 E Z 1 2 6 Z Z-6-Metilhept-2-eno (2Z,4E)-Hexa-2,4-dieno
 delante del nombre. Si hubiese varios, se escriben entre paréntesis y se indica su posición con localizadores. En caso de igualdad Z precede a E E. Z Z. Z-6-Metilhept-2-eno. (2Z,4E)-Hexa-2,4-dieno.")
91
3. 5. Radicales monovalentes: se cambia -eno por –enilo
3.5. Radicales monovalentes: se cambia -eno por –enilo y -ino por –inilo. El carbono 1 del radical es el unido a la cadena principal. 1 4 7 8 1’ 2’ 3’ PROPINO 2-PROPINILO 2-PROPINIL (PROP-2-INIL) 4-(2-Propinil)-octa-1,7-dieno 4-2’-Propinilocta-1,7-dieno 4-Propargilocta-1,7-dieno
 4-(2-Propinil)-octa-1,7-dieno. 4-2’-Propinilocta-1,7-dieno. 4-Propargilocta-1,7-dieno.")
92
3. 6. Radicales bi- y trivalentes: se cambia la o- del
3.6. Radicales bi- y trivalentes: se cambia la o- del radical saturado por -ideno o -idino 1 3 4 6 8 METILO METILIDENO METILIDEN 3-Etil-4-metilidenocta-1,6-dieno

93
Hidrocarburos Alifáticos Cíclicos:
Cicloalcanos, cicloalquenos y cicloalquinos

94
1. 1. Se antepone el prefijo ciclo- al nombre del alcano
1.1. Se antepone el prefijo ciclo- al nombre del alcano de igual número de carbonos Cicloalcano Propano Ciclopropano Ciclohexano Ciclooctano

95
1.2. Los radicales se nombran cambiando -ano por -ilo
Ciclopropilo Ciclopropano Ciclohexilo

96
1. 3. Cicloalcanos sustituidos: Se utilizan las mismas reglas
1.3. Cicloalcanos sustituidos: Se utilizan las mismas reglas que para alcanos. Cuando sólo hay un sustituyente, no se precisa localizador. 1-Etil-2-metilciclopentano Isopropilciclohexano 1.4. Cicloalquenos y cicloalquinos: Se utilizan las mismas reglas que para alquenos y alquinos 5-Metilciclohexa-1,3-dieno 1 3 5 Metilidenciclopentano 3-Metilciclohexeno

97
ISOMERÍA EN CICLOALCANOS
cis-1,2-Dimetilciclohexano 1,2-Dimetilciclohexano trans-1,2-Dimetilciclohexano

98
1.5. Algunos ejemplos de cicloalcanos unidos por enlace C-C
A) Con ciclos iguales: 1 1’ 1’ 1 1,1’-Biciclohexilo 1,1’-Biciclohexano Biciclohexano 1,2’-Dimetil-1,1’-biciclobutilo 1,2’-Dimetil-1,1’-biciclobutano
 Con ciclos iguales: 1. 1’ 1’ 1. 1,1’-Biciclohexilo. 1,1’-Biciclohexano. Biciclohexano. 1,2’-Dimetil-1,1’-biciclobutilo. 1,2’-Dimetil-1,1’-biciclobutano.")
99
B) Con ciclos diferentes:
se toma como base el mayor de ellos y el resto se nombran como sustituyentes en orden alfabético ANILLO PRINCIPAL localizadores en orden alfabético 1 3 2 Ciclobutilciclopentano 1-Etil-3(2-metilciclopentil)ciclohexano 2 1 5 5-Ciclobutil-1-ciclopentil-2-ciclopropilcicloheptano
ciclohexano Ciclobutil-1-ciclopentil-2-ciclopropilcicloheptano.")
100
Biciclos y Espiranos

101
Carbonos cabeza de puente Comunes a varios puentes
1.1. Biciclos: Entre dos carbonos no contiguos del anillo tienen un enlace, un átomo o una cadena. Carbonos del puente más largo Carbonos cabeza de puente Comunes a varios puentes Carbonos del puente más corto Carbonos del puente intermedio Biciclo[3.2.1]octano Sustituyentesbiciclo [..] nombre de alcano de igual nº de C En orden decreciente Biciclo[4.2.0]octano Biciclo[3.1.1]heptano

102
1. 2. Numeración: El carbono 1 es un C cabeza de puente
1.2. Numeración: El carbono 1 es un C cabeza de puente. Se numera hacia el otro cabeza de puente por el puente más largo, luego el puente intermedio y finalmente el más corta. En caso de igualdad se aplican localizadores más bajos a los sustituyentes. 2-Metilbiciclo[2.2.1]heptano E antes que M > = 2-Etil-4,6,8-trimetilbiciclo[3.2.2]nonano

103
2.Espiranos: Dos ciclos que tienen sólo un carbono común
Carbonos pertenecientes sólo al ciclo grande Carbono espiránico Carbonos pertenecientes sólo al ciclo pequeño Espiro [2.5] octano NOMBRE 1: localizadores-sustituyentes + espiro [. ] alcano de igual nº de C Numeración: Se empieza por el ciclo pequeño dando el nº 1 a un carbono contiguo al espiránico, y se continua por el grande pasando por el carbono espiránico 4-Etil-1-metilespiro [2.5] octano

104
Ciclohexanoespirociclopropano
NOMBRE 2: localizadores-sustituyentes + cicloalcano mayor espiro + cicloalcano menor Ciclohexanoespirociclopropano Numeración: Empezando por el carbono espiránico se numera el ciclo grande completo. A continuación, y empezando por el carbono espiránico se numera el ciclo pequeño completo con múmeros primados 2-Etil-2’-metilciclohexanoespirociclopropano

105
Hidrocarburos aromáticos

106
Nombre: localizadores + sustituyentes + benceno
1.1. Mononucleares Nombre: localizadores + sustituyentes + benceno Monosustituídos No necesita localizador 6 carbonos equivalentes Vinilbenceno Polisustituídos Numeración: Se dan los nos más bajos a los sustituyentes 2-Etil-1-metil-4-propilbenceno

107
Ph- Ar- 1.2. Nombres Propios y Radicales Fenilo Arilo Disustituídos
orto- meta- para- o- m- p- 1,2- 1,3- 1,4-

108
1.3. Polinucleares Posiciones equivalentes Naftaleno Antraceno 9 y 10
1,4,5 y 8 (a) 2,3,6 y 7 (b) Antraceno 1,4,5 y 8 (a) 2,3,6 y 7 (b) 9 y 10 Fenantreno 1 y 8 (a) 2 y 7 () 3 y 6 4 y 5 9 y 10
 2,3,6 y 7 (b) Antraceno. 1,4,5 y 8 (a) 2,3,6 y 7 (b) 9 y 10. Fenantreno. 1 y 8 (a) 2 y 7 () 3 y 6. 4 y 5. 9 y 10.")
109
Tema 4-Las Reacciones Orgánicas
QUIMICA ORGANICA Tema 4-Las Reacciones Orgánicas

110
PRINCIPALES TIPOS DE REACCIONES ORGÁNICAS
REACCIONES DE SUSTITUCIÓN REACCIONES DE ADICIÓN REACCIONES DE ELIMINACIÓN REACCIONES DE TRANSPOSICIÓN REACCIONES DE CONDENSACIÓN

111
MECANISMOS DE REACCIÓN
REACCIONES HOMOLÍTICAS Y HETEROLÍTICAS REACCIONES HOMOLÍTICAS REACCIONES HETEROLÍTICAS

112
REACTIVOS NUCLEÓFILOS Y ELECTRÓFILOS
REACCIONES HETEROLÍTICAS Nucleófilo Electrófilo Nucleófilo Electrófilo

113
ÁCIDOS Y BASES ORGÁNICOS

114
PERFIL ENERGÉTICO DE UNA REACCIÓN
REACCIONES CONCERTADAS REACCIONES NO CONCERTADAS O REACCIONES POR PASOS

115
Carbaniones Radicales libres Carbocationes
ESTRUCTURA Y ESTABILIDAD RELATIVA DE LOS PRINCIPALES INTERMEDIOS DE REACCIÓN Radicales libres Carbaniones Carbocationes

116
ESTABILIDAD RELATIVA DE RADICALES LIBRES
Conjugación con e- . Hiperconjugación con e- de enlaces . Radical alilo

117
Radical etilo Radical etenilo o vinilo Radical fenilo Radical etinilo

118
ESTABILIDAD RELATIVA DE CARBOCATIONES
Conjugación con e- . Hiperconjugación con e- de enlaces . Efecto inductivo (+I). Catión alilo
. Catión alilo.")
119
Catión etilo Catiónl etenilo o vinilo Catión fenilo Catiónl etinilo

120
ESTABILIDAD RELATIVA DE CARBANIONES
Sistemas alquílicos sencillos: muy inestables. Factores que estabilizan carbaniones: Aumento del carácter s del C que soporta la carga negativa.

121
Conjugación del par electrónico con un doble enlace C=C.
Anión alilo

122
Conjugación del par electrónico con enlaces múltiples C-Heteroátomo.
Grupos con efecto -I unidos al C aniónico Deslocalización de la carga negativa en orbitales d vacantes de átomos contiguos al C aniónico.

123
POLARIDAD DE LOS ENLACES COVALENTES
ENLACE COVALENTE PURO ENLACE COVALENTE POLAR (parcialmente iónico) H - F
 H - F.")
124
Electronegatividades de Pauling de algunos elementos comunes
en los compuestos orgánicos H 2.2 Li 1.0 Be 1.6 B 1.8 C 2.5 N 3.0 O 3.4 F 4.0 Na 0.9 Mg 1.3 Al Si 1.9 P S 2.6 Cl 3.3 K 0.8 Br I 2.7

125
Definición de Dipolo - + m = q .d El dipolo es una entidad física
constituída por un par de cargas eléctricas (polos) de igual valor absoluto y signo contrario situadas a una distancia finita (d). Momento dipolar + - d q+ = q- Eje del dipolo Centro del dipolo El módulo es igual al producto de la carga por la distancia = q .d m
 de igual valor absoluto. y signo contrario situadas a una distancia. finita (d). Momento dipolar. + - d. q+ = q- Eje del dipolo. Centro del dipolo. El módulo es igual al producto. de la carga por la distancia. = q .d. m.")
126
Momento dipolar molecular

127
EFECTOS ELECTRÓNICOS EFECTO INDUCTIVO
Polarización de los enlaces producida por un átomo o grupo atómico. Disminuye rápidamente con la distancia. Los electrones no cambian de orbital. Es una propiedad intrínseca de la molécula. Supone una corrección a las fórmulas clásicas.

130
EFECTO CONJUGATIVO. RESONANCIA
Hace referencia a la descripción de una molécula mediante dos o más fórmulas que solo difieren en la posición de los electrones Fórmulas canónicas o Formas resonantes

131
EFECTO CONJUGATIVO Hace referencia a la corrección de las fórmulas clásicas en las que existen enlaces múltiples y sistemas conjugados. Intervienen electrones de sistemas múltiples (p ) o pares de electrones sin compartir (n). Cambia la estructura electrónica de los átomos. Los electrones cambian de orbital. Se transmite integramente a lo largo de un sistema conjugado.
 o pares de electrones. sin compartir (n). Cambia la estructura electrónica de los átomos. Los electrones. cambian de orbital. Se transmite integramente a lo largo de un sistema conjugado.")
133
EFECTOS INDUCTIVOS Y CONJUGATIVOS DE ALGUNOS GRUPOS FUNCIONALES
+I, +R +R >> -I +R > -I +I -I > +R -R , -I -I +R o -R

134
EFECTO HIPERCONJUGATIVO
RESONANCIA DE NO ENLACE. Hace referencia a la corrección de las fórmulas clásicas mediante la participación de electrones de enlaces s. Cambia la estructura electrónica de los átomos. Los electrones cambian de orbital.

135
REGLAS PARA FORMULAR ESTUCTURAS RESONANTES
Todos los átomos deben ocupar las mismas posiciones en todas las estructuras resonantes. Todas las estructuras deben presentar el mismo número de electrones en el mismo estado de apareamiento. Para establecer el orden de estabilidad se deben tener en cuenta los siguientes puntos: 1. Las estructuras en las que los átomos tienen su capa de valencia completa (regla del octete) son más estables.
 son más estables.")
136
2. Las estructuras son tanto más estables cuanto mayor sea su
carácter covalente (mayor número de enlaces formales). 3. En general, las fórmulas neutras son más estables que las que presentan separación de cargas. 4. Entre las estructuras con cargas, y en igualdad de otras condiciones, será más importante la estructura que sitúa las cargas de acuerdo con la electronegatividad de los átomos.
. 3. En general, las fórmulas neutras son más estables que las que. presentan separación de cargas. 4. Entre las estructuras con cargas, y en igualdad de otras condiciones, será más importante la estructura que sitúa las cargas de acuerdo con. la electronegatividad de los átomos.")
137
Excepción: la regla del octete tiene prioridad sobre el criterio de
electronegatividad. 5. Las estructuras resonantes en las que sea necesario situar pares de electrones p o pares de electrones sin compartir en orbitales con n=3 para elementos del primer período largo del Sistema Periódico (C, N, O) presentan una contribución despreciable. Las estructuras resonantes equivalentes tienen el mismo contenido de energía.
 presentan una contribución despreciable. Las estructuras resonantes equivalentes tienen el mismo contenido de. energía.")
138
Deslocalización electrónica ER
La energía de resonancia (ER) aumenta al: aumentar el número de estructuras resonantes aumentar la analogía de las estructuras resonantes que participan disminuir el contenido de energía de las estructuras equivalentes. Deslocalización electrónica ER Estabilización
 aumenta al: aumentar el número de estructuras resonantes. aumentar la analogía de las estructuras resonantes. que participan. disminuir el contenido de energía de las estructuras. equivalentes. Deslocalización electrónica ER. Estabilización.")
139
FUERZAS INTERMOLECULARES
Son de naturaleza electrostática, pero mucho más débiles que las fuerzas de enlace. Interacciones ión -dipolo Interacciones atractivas entre las moléculas neutras Interacciones dipolo-dipolo Interacciones de dispersión de London Interacciones de puente de hidrógeno Típicamente las interacciones dipolo-dipolo y de dispersión se agrupan y se denominan como fuerzas de van der Waals.

140
Interacciones ión -dipolo
Se trata de una interacción entre un ión cargado y una molécula polar (un dipolo) Los cationes se verán atraídos por el lado negativo de un dipolo Los aniones se verán atraídos por el lado positivo de un dipolo Este tipo de interacciones tienen gran importancia al estudiar el comportamiento de las disoluciones de sustancias iónicas en disolventes polares (por ejemplo sal en agua).
 Los cationes se verán atraídos por el lado negativo de un dipolo. Los aniones se verán atraídos por el lado positivo de un dipolo. Este tipo de interacciones tienen gran importancia al estudiar el comportamiento de las disoluciones de sustancias iónicas en disolventes polares (por ejemplo sal en agua).")
141
Interacciones dipolo-dipolo
FUERZAS DIPOLO-DIPOLO existen entre moléculas polares neutras Son efectivas cuando las moléculas están muy próximas • Son más débiles que las ión-dipolo Las fuerzas dipolo-dipolo aumentan con la polaridad de la molécula

142
Interacciones de dispersión de London
Interacciones dipolo-dipolo inducido Fuerzas de atracción existentes entre moléculas no polares, mediante dipolos instantáneos generados por el movimiento de los electrones (polarizabilidad). Un dipolo temporal puede inducir un dipolo similar en un átomo vecino, si este está lo suficientemente cercano Estas interacciones son significativas únicamente cuando los átomos o las moléculas están muy próximas
. Un dipolo temporal puede inducir un dipolo similar. en un átomo vecino, si este está lo suficientemente cercano. Estas interacciones son significativas únicamente cuando los átomos o las. moléculas están muy próximas.")
143
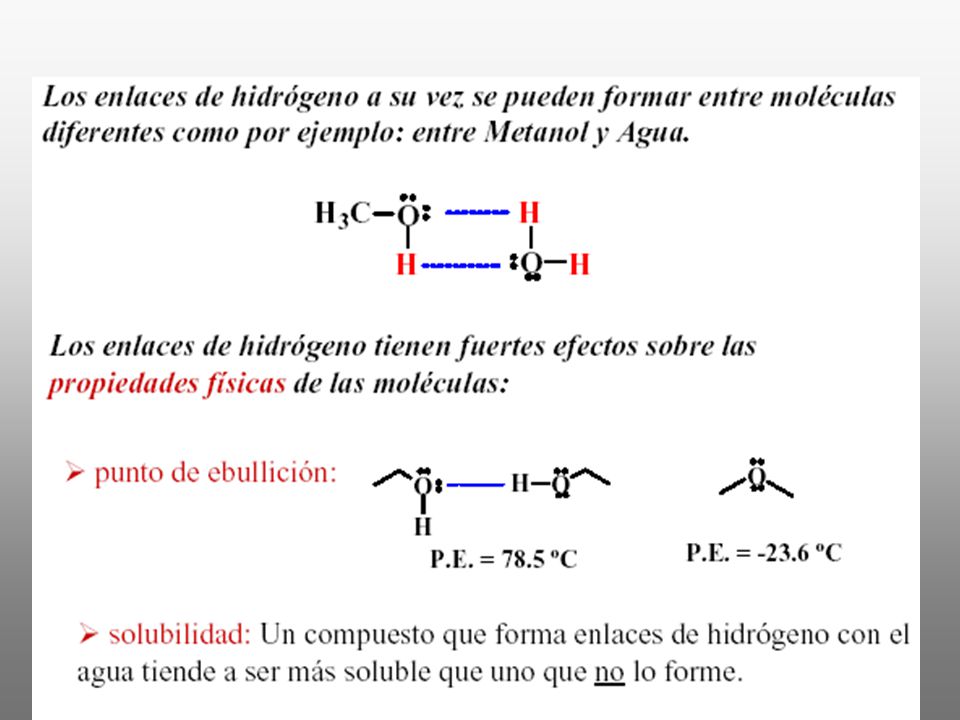
Enlace o puentes de hidrógeno
Puente de hidrógeno es la atracción inter o intramolecular resultante de la interacción de dipolos donde participa el H. Un enlace entre un hidrógeno y un átomo electronegativo como el F, O o N es muy polar: Un átomo de hidrógeno en un enlace polar (por ejemplo H-F, H-O o H-N) puede experimentar una fuerza atractiva hacia una molécula polar o un ion vecino que cuenta con pares electrónicos sin compartir, usualmente átomos de F, O o N Esta atracción es responsable de muchas de las propiedades características del H2O, Estas interacciones son más fuertes que las dipolo-dipolo y de London.
 puede experimentar una fuerza atractiva hacia una molécula polar o un ion vecino que cuenta con pares electrónicos sin compartir, usualmente átomos de F, O o N. Esta atracción es responsable de muchas de. las propiedades características del H2O, Estas interacciones son más fuertes que las. dipolo-dipolo y de London.")
145
Tema 7-Hidrocarburos con Dobles Enlaces: ALQUENOS Y CICLOALQUENOS

146
Estructura Los alquenos son hidrocarburos con dobles enlaces carbono-carbono. Se denominan también olefinas. Estas distancias y ángulos de enlace se pueden explicar admitiendo que los dos átomos de carbono que forman el doble enlace presentan una hidridación sp2 y que el doble enlace está constituido por un enlace y un enlace π. La energía de disociación del doble enlace C=C es aproximadamente de 146 kcal/mol y la energía de disociación de un enlace sencillo C-C es de 83 kcal/mol. Por tanto, la energía de disociación del enlace p debe ser de 63 kcal/mol.

147
Estereoisomeria en Alquenos
ISOMERÍA GEOMÉTRICA A diferencia de lo que ocurre en los enlaces sencillos, el giro en torno a un doble enlace carbono-carbono está muy impedido. Es necesario superar una barrera de energía elevada, porque para ello se debería romper el enlace . Este es el origen de estereoisómeros (diastereómeros) en alquenos, que se suelen denominar isómeros geométricos y se diferencian en la disposición espacial de los grupos en torno al doble enlace. NOMENCLATURA Cis-Trans
 en alquenos, que se. suelen denominar isómeros geométricos y se diferencian en la disposición. espacial de los grupos en torno al doble enlace. NOMENCLATURA Cis-Trans.")
148
NOMENCLATURA Z/E: Z (zusammen = juntos), E (entgegen = opuestos).
(E)-2,5-dimetilhex-3-eno (2E,4Z)-2,4-heptadieno
-2,5-dimetilhex-3-eno. (2E,4Z)-2,4-heptadieno.")
149
REACTIVIDAD QUÍMICA Como el enlace C-C es más estable que el enlace es de esperar que los alquenos reaccionen dando compuestos con enlaces sencillos de tipo . Reacciones de Adición Mientras que los electrones del enlace están fuertemente unidos en el doble enlace C=C, la densidad electrónica que forma el enlace está deslocalizada por arriba y por abajo del enlace . Los electrones del enlace están colocados lejos de los núcleos de carbono y unidos con menos fuerza a éstos: la nube electrónica es más deformable (más polarizable) por la acción de agentes externos. REACCIONES DE ADICIÓN ELECTRÓFILA AdE REACCIONES DE ADICIÓN vía radicálica HIDROGENACIÓN REACCIONES DE OXIDACIÓN
 por la acción de agentes externos. REACCIONES DE ADICIÓN ELECTRÓFILA AdE. REACCIONES DE ADICIÓN vía radicálica. HIDROGENACIÓN. REACCIONES DE OXIDACIÓN.")
150
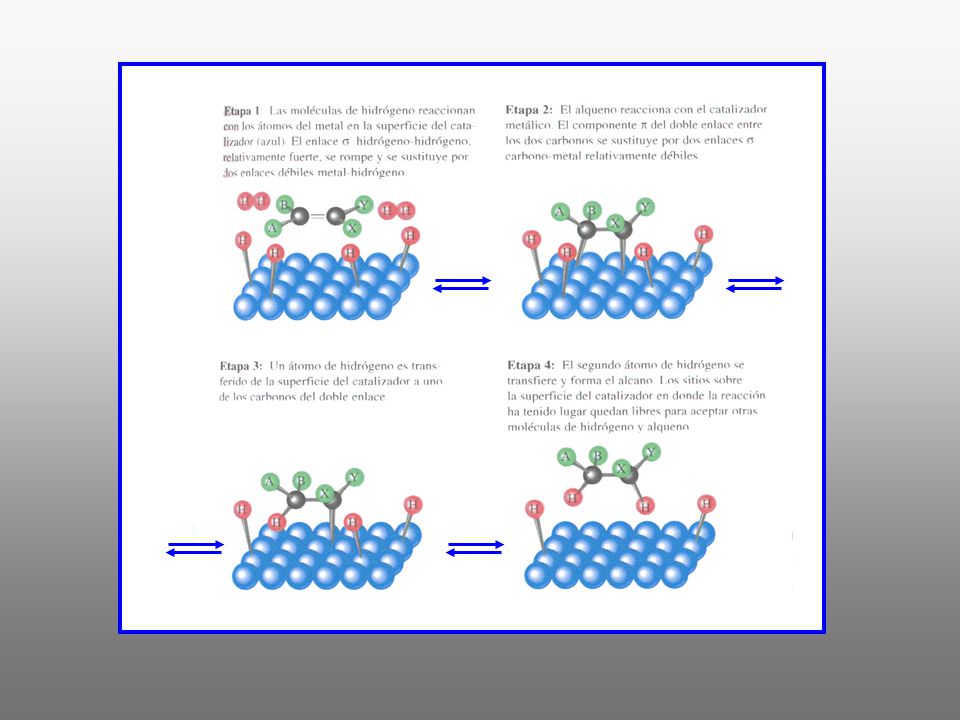
HIDROGENACIÓN La hidrogenación de un alqueno consiste en la adición de H2 al doble enlace para dar un alcano. Para que la reacción tenga lugar a una velocidad adecuada se emplea un catalizador Catalizadores: metales finamente divididos Pt, Pd Rh, Ru,Ni. Sistemas catalíticos. Metal/Soporte (carbón, CaCO3, BaSO4): Pd/C Disolventes: un alcohol, un alcano o ácido acético. La reacción se efectúa disolviendo el alqueno en el disolvente elegido, agregando una pequeña cantidad de catalizador y agitando la mezcla en una atmósfera de hidrógeno. La hidrogenación tiene lugar en la superficie del catalizador metálico CATÁLISIS HETEROGÉNEA
: Pd/C. Disolventes: un alcohol, un alcano o ácido acético. La reacción se efectúa disolviendo el alqueno en el disolvente elegido, agregando una. pequeña cantidad de catalizador y agitando la mezcla en una atmósfera de hidrógeno. La hidrogenación tiene lugar en la superficie del catalizador metálico. CATÁLISIS HETEROGÉNEA.")
152
CARACTERÍSTICAS *Cuanto menor es el número y tamaño de los sustituyentes del doble enlace mayor es la velocidad de reacción. CH2=CH2 > R-CH=CH2 > R-CH=CH-R > R2C=CH-R > R2C=CR2 * La reacción transcurre con una estereoquímica SUPRAFACIAL, SUPRA o SIN * La reacción es ESTEREOSELECTIVA

153
Estabilidades relativas de los alquenos
Las estabilidades relativas de los alquenos se pueden establecer comparando los calores de hidrogenación (H°). La hidrogenación es ligeramente exotérmica
. La hidrogenación es ligeramente exotérmica.")
154
REACCIONES DE ADICIÓN ELECTRÓFILA

155
REACCIONES DE ADICIÓN ELECTRÓFILA DE ÁCIDOS
ADICIÓN DE HALUROS DE HIDRÓGENO

156
Orientación de la adición: Regla de Markovnikov.
En 1869 Markovnikov demostró que la orientación de la adición de HBr a los alquenos era regioselectiva y postuló la siguiente norma conocida como regla de Markovnikov: En una reacción de adición iónica de un ácido (HX) a un alqueno no simétrico el producto principal es el que procede de la unión del protón (H) al carbono del doble enlace que contiene mayor número de átomos de hidrógeno (más hidrogenado). Justificación En función del mecanismo de la reacción el paso clave del proceso es la formación del carbocatión y de este modo se forma como intermedio el carbocatión más estable
 a un alqueno no simétrico el producto. principal es el que procede de la unión del protón (H) al carbono del doble enlace que. contiene mayor número de átomos de hidrógeno (más hidrogenado). Justificación. En función del mecanismo de la reacción. el paso clave del proceso es la formación del carbocatión y de este modo se forma. como intermedio el carbocatión más estable.")
157
Regla de Markovnikov generalizada:
En las reacciones de adición electrófila a alquenos no simétricos el producto principal se forma por unión del extremo electrófilo del reactivo al átomo de carbono más hidrogenado o, lo que es equivalente, por unión del extremo nucleófilo del reactivo al carbono que mejor soporta la carga positiva en el catión intermedio.

159
Adición de HBr a alquenos en presencia de peróxidos
(mecanismo radicálico) P- Markovnikov P. Anti-Markovnikov Mecanismo
 P- Markovnikov. P. Anti-Markovnikov. Mecanismo.")
160
el isómero anti-Markovnikov. Por ejemplo,
Este mecanismo explica por qué la adición radicálica de HBr a olefinas proporciona el isómero anti-Markovnikov. Por ejemplo, 2ª. Etapa de propagación

161
La reacción por via radicálica ocurre con HBr pero no con HCl o HI.
Justificación: Solo en la reacción con HBr los dos pasos de la etapa de propagación son exotérmicos y poseen bajas energías de activación. Las reacciones con otros ácidos fuertes como H2SO4, CF3COOH, transcurren de forma similar (AdE).
.")
162
REACCIONES DE HIDRATACIÓN
Cuando un alqueno reacciona con agua en presencia de un catalizador fuertemente ácido se obtiene un alcohol. A este proceso se le denomina reacción de hidratación de alquenos porque formalmente se agregan los elementos del agua (un átomo de hidrógeno H y un grupo hidroxilo OH) al doble enlace. se emplean como catalizadores ácidos fuertes no nucleofílicos, como H2SO4 o H3PO4. La reacción es un equilibrio (exceso de agua) Mecanismo
 al doble enlace. se emplean como catalizadores ácidos fuertes no nucleofílicos, como H2SO4 o H3PO4. La reacción es un equilibrio (exceso de agua) Mecanismo.")
163
Muchos alquenos no se hidratan fácilmente en ácidos acuosos diluidos debido a que son poco
solubles en el medio de reacción o el equilibrio está desplazado hacia el alqueno. Para favorecer el proceso de hidratación con una orientación Markovnikov se puede emplear la siguiente secuencia: Tratamiento con ácido sulfúrico seguida de hidrólisis del hidrógenosulfato de alquilo.

164
REACCIONES DE ADICIÓN ELECTRÓFILA DE ÁCIDOS: CARACTERÍSTICAS COMUNES
* Transcurren a través de carbocationes (intermedios de cadena abierta). * El orden de reactividad de los alquenos es función de la estabilidad del carbocatión intermedio. El alqueno más reactivo es el que puede formar el carbocatión más estable. * Orientación o Regioselectividad: Regla de Markovnikov. * Las reacciones no son estereoselectivas. * Transposiciones( Wagner-Meerwein)
. * El orden de reactividad de los alquenos es función de la estabilidad del. carbocatión intermedio. El alqueno más reactivo es el que puede formar. el carbocatión más estable. * Orientación o Regioselectividad: Regla de Markovnikov. * Las reacciones no son estereoselectivas. * Transposiciones( Wagner-Meerwein)")
165
HALOGENACIÓN La molécula de halógeno, al estar formada por dos átomos idénticos y por tanto de igual electronegatividad, no está polarizada. Sin embargo, cuando se aproxima a la nube del alqueno sufre una polarización temporal dando lugar a la formación de un complejo . El complejo . evoluciona para dar un ión halogenonio cíclico, por unión del X+, y un ión haluro (X-). Mecanismo iónico AdE (disolvente inerte, ausencia de luz, temperatura ambiente) Ión bromonio Intermedio
. Mecanismo iónico AdE (disolvente inerte, ausencia de luz, temperatura ambiente) Ión bromonio. Intermedio.")
166
El ión halogenonio experimenta el ataque del ión haluro (nucleófilo), para dar el
producto dihalogenado * La reacción transcurre con una estereoquímica ANTARAFACIAL, ANTARA o ANTI * La reacción es ESTEREOSELECTIVA

167
Mecanismo radicálico: reacción en cadena
(1S2S) (1R2R) Mecanismo radicálico: reacción en cadena (en presencia de luz, o por via térmica) 2ª. Etapa de propagación * La reacción no es ESTEREOSELECTIVA 1,2-dibromo-2-metilpropano
 (1R2R) Mecanismo radicálico: reacción en cadena. (en presencia de luz, o por via térmica) 2ª. Etapa de propagación. * La reacción no es ESTEREOSELECTIVA. 1,2-dibromo-2-metilpropano.")
168
Reacción de sustitución alílica
2ª. Etapa de propagación 1,2-dibromo-2-metilpropano Favorecida aT y concentraciones de halógeno

169
FORMACIÓN DE HALOHIDRINAS
Cuando un alqueno reacciona con un halógeno (Cl2, Br2) en presencia de agua, o con HOCl, HOBr el producto de la reacción contiene un átomo de halógeno y un grupo hidroxilo en átomos de carbono adyacentes. Estos compuestos se denominan genéricamente halohidrinas (bromohidrina, clorohidrina). El mecanismo del proceso es similar al de la reacción de halogenación, pero con la diferencia de que el nucleófilo del proceso es el H2O, en lugar de un ión haluro. * Orientación o Regioselectividad: Regla de Markovnikov generalizada * La reacción transcurre con una estereoquímica ANTARAFACIAL, ANTARA o ANTI * La reacción es ESTEREOSELECTIVA
 en presencia de agua, o con. HOCl, HOBr el producto de la reacción contiene un átomo de halógeno y un grupo. hidroxilo en átomos de carbono adyacentes. Estos compuestos se denominan. genéricamente halohidrinas (bromohidrina, clorohidrina). El mecanismo del proceso es similar al de la reacción de halogenación, pero con la. diferencia de que el nucleófilo del proceso es el H2O, en lugar de un ión haluro. * Orientación o Regioselectividad: Regla de Markovnikov generalizada. * La reacción transcurre con una estereoquímica ANTARAFACIAL, ANTARA o ANTI. * La reacción es ESTEREOSELECTIVA.")
170
Ión bromonio Intermedio

171
HIDROBORACIÓN * Orientación o Regioselectividad: Regla de Markovnikov generalizada * La reacción transcurre con una estereoquímica SUPRAFACIAL, SUPRA o SIN * La reacción es ESTEREOSELECTIVA

172
LOS ALQUILBORANOS SON IMPORTANTES INTERMEDIOS EN SÍNTESIS
* Descomposición oxidativa: conduce a alcoholes. El proceso global: hidroboración + desomposición oxidativa supone la adición de H2O con una orientación Anti-Markovnikov y una estereoquímica SUPRAFACIAL * Hidrólisis ácida: conduce a alcanos. El proceso global: hidroboración + hidrólisis ácida supone la adición de H2 con una estereoquímica SUPRAFACIAL

173
OXIMERCURIACIÓN-DESMERCURIACIÓN
El proceso global conduce a un alcohol y supone la adición de H2O con una orientación Markovnikov. P- Markovnikov P. Anti-Markovnikov

174
HIDROXILACIÓN DE ALQUENOS
OXIDACIÓN DE ALQUENOS HIDROXILACIÓN DE ALQUENOS El tetróxido de osmio (OsO4) se adiciona al doble enlace de los alquenos para formar un osmiato cíclico, que posteriormente por hidrólisis reductora conduce a un 1,2-diol (glicol) La ruptura y formación de enlaces en el proceso de adición del OsO4 tiene lugar de forma concertada. Los dos enlaces carbono-oxígeno se forman simultáneamente y por la misma cara del doble enlace. * La reacción transcurre con una estereoquímica SUPRAFACIAL, SUPRA o SIN * La reacción es ESTEREOSELECTIVA
 se adiciona al doble enlace de los alquenos para. formar un osmiato cíclico, que posteriormente por hidrólisis reductora conduce a. un 1,2-diol (glicol) La ruptura y formación de enlaces en el proceso de adición del OsO4 tiene lugar. de forma concertada. Los dos enlaces carbono-oxígeno se forman simultáneamente y. por la misma cara del doble enlace. * La reacción transcurre con una estereoquímica SUPRAFACIAL, SUPRA o SIN. * La reacción es ESTEREOSELECTIVA.")
175
La hidroxilación de alquenos también se puede conseguir mediante la reacción con una
disolución acuosa básica diluida y fría de permanganato potásico (KMnO4). El anión permanganato se adiciona al doble enlace mediante un mecanismo similar al del OsO4, formando un éster cíclico que por hidrolisis en el medio básico conduce a un 1,2-diol.
. El anión. permanganato se adiciona al doble enlace mediante un mecanismo similar al del OsO4, formando un éster cíclico que por hidrolisis en el medio básico conduce a un 1,2-diol.")
176
EPOXIDACIÓN DE ALQUENOS
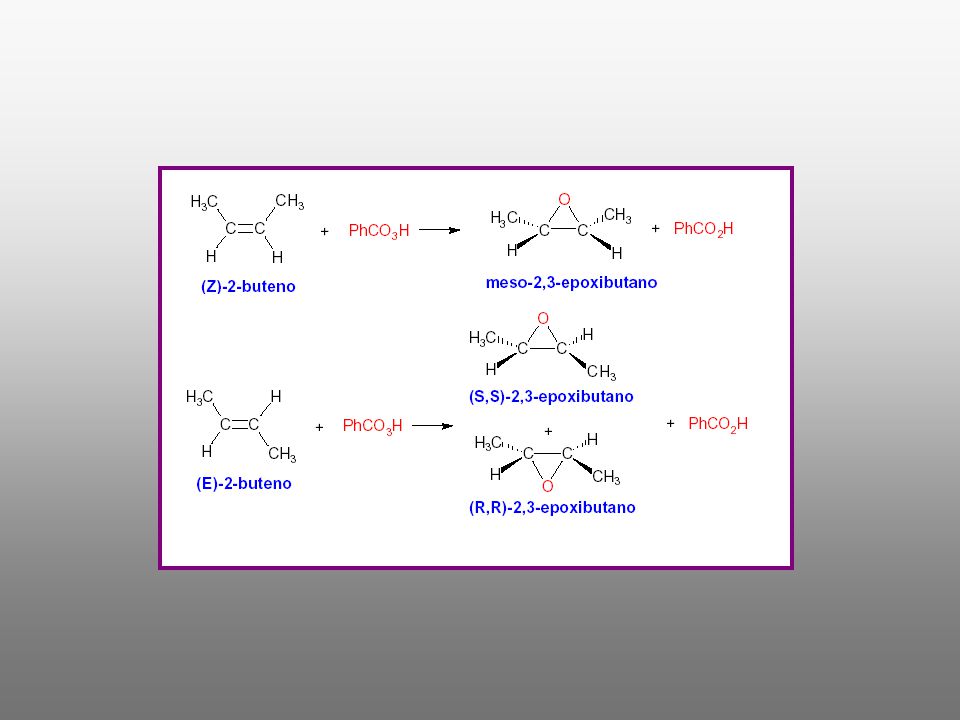
Un epóxido, llamado también oxirano, es un éter cíclico de tres eslabones. Los reactivos que permiten transformar los alquenos en epóxidos son los peroxiácidos (perácidos): ácidos carboxílicos con un átomo adicional de oxígeno en un enlace peroxi –O-O-. Como la reacción de epoxidación tiene lugar en un solo paso la estereoquímica presente en el alqueno se retiene en el epóxido. * La reacción transcurre de forma concertada con una estereoquímica SUPRAFACIAL, SUPRA o SIN * La reacción es ESTEREOSELECTIVA
: ácidos carboxílicos con un átomo adicional de oxígeno en un enlace. peroxi –O-O-. Como la reacción de epoxidación tiene lugar en un solo paso la estereoquímica. presente en el alqueno se retiene en el epóxido. * La reacción transcurre de forma concertada con una estereoquímica. SUPRAFACIAL, SUPRA o SIN. * La reacción es ESTEREOSELECTIVA.")
178
Hidrólisis básica de Oxiranos
El tratamiento de un oxirano o epóxido con HO-/H2O conduce a un 1,2-diol. El ataque del anión HO- tiene lugar con una estereoquímica ANTARAFACIAL. La secuencia epoxidación + hidrólisis básica conduce a 1,2-dioles con una estereoquímica ANTI Hidrólisis ácida de Oxiranos El tratamiento de un oxirano o epóxido con H+/H2O conduce a un 1,2-diol.

179
RUPTURA OXIDATIVA DE ALQUENOS
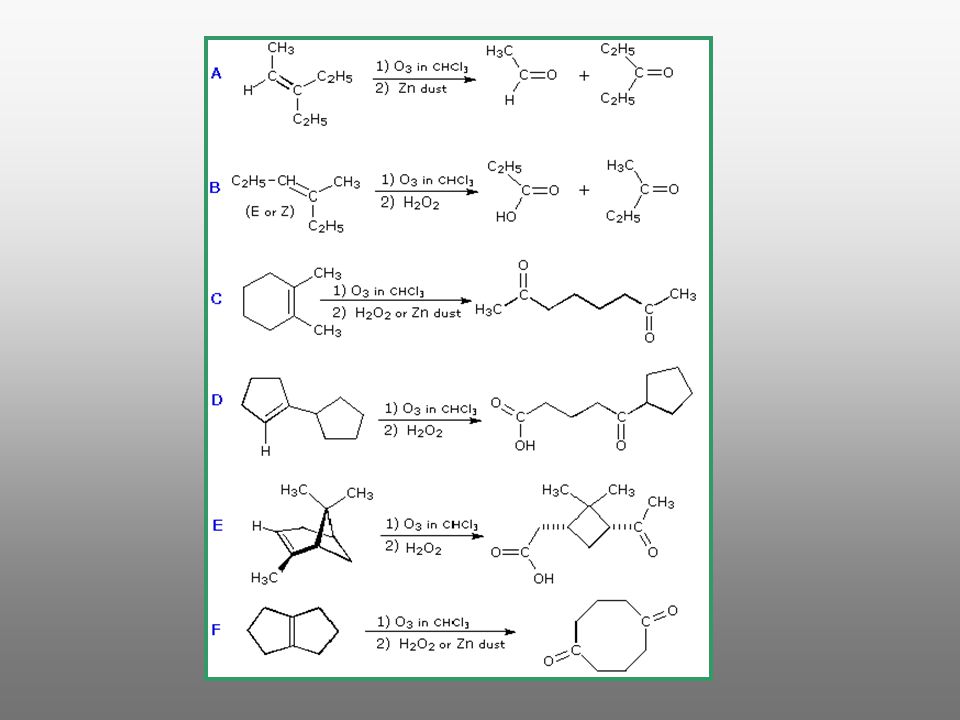
Método de interés para: * La síntesis de oxocompuestos y/o ácidos carboxílicos * la determinación de la estructura de alquenos 1. Empleando condiciones de oxidación enérgicas por ejemplo KMnO4 en medio ácido o básico / D K2Cr2O7 / H2SO4 / D 2. Ozonolísis

180
OZONOLÍSIS El ozono se forma cuando la luz ultravioleta o una descarga eléctrica pasan a través de oxígeno gaseoso. La luz ultravioleta de origen solar convierte al oxígeno de las capas altas de la atmósfera en ozono. En la estratosfera el ozono se forma constantemente a partir del oxígeno mediante la absorción de la radiación ultravioleta que llega del sol. En un proceso de equilibrio, el ozono vuelve a absorber radiación solar y se rompe generando oxígeno. El ozono es mucho más reactivo que el oxígeno porque su contenido energético supera en 32 kcal/mol al del oxígeno. El ozono se puede describir mediante un híbrido de resonancia

181
Los alquenos reaccionan con el ozono para formar un compuesto cíclico denominado
molozónido. El molozónido tiene dos enlaces peróxido y es bastante inestable y se transpone inmediatamente, aun a muy bajas temperaturas, para generar un compuesto denominado ozónido

185
CICLOPROPANACIÓN Reacción de Simmons-Smith

186
APLICACIONES SINTÉTICAS

187
p Tema 8. Sistemas -deslocalizados
Polienos: clasificación estructural y estabilidad relativa Dienos aislados Dienos conjugados Dienos acumulados Alenos

188
Dienos conjugados: estructura, estabilidad y reactividad
HOMO LUMO

189
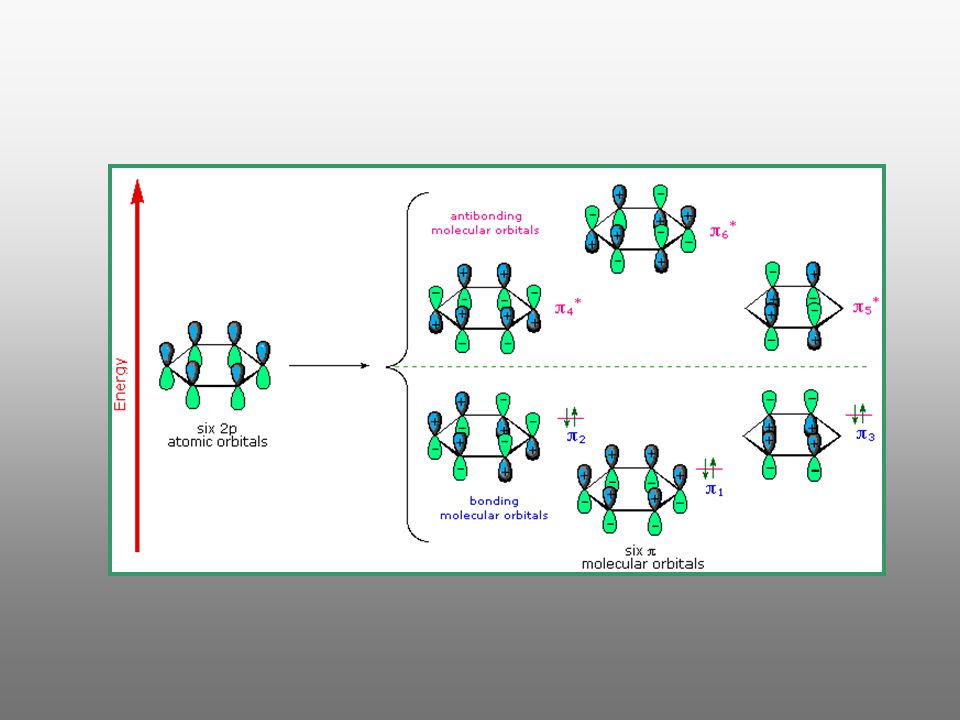
Conjugación extendida en el benceno. Aromaticidad

192
REACCIONES DE ADICIÓN ELECTRÓFILA : ADICIÓN 1,2 Y 1,4

194
SÍNTESIS DEL FUNGICIDA CAPTÁN

Presentaciones similares
















 Reactividad de los compuestos de carbono>")



>")












 Reactividad de los compuestos de carbono>")
