Descargar la presentación
La descarga está en progreso. Por favor, espere

1
Hiperbilirrubinemias Hereditarias
Química Biológica Patológica Hiperbilirrubinemias Hereditarias Tema 4- Bolilla 4 Dra. Silvia Varas

2
METABOLISMO DE BILIRRUBINA

4
2 3 1 4 +H+ A B D C 8 5 7 6 A B +H+ D C

8
ELIMINACIÓN DE LA BILIRRUBINA
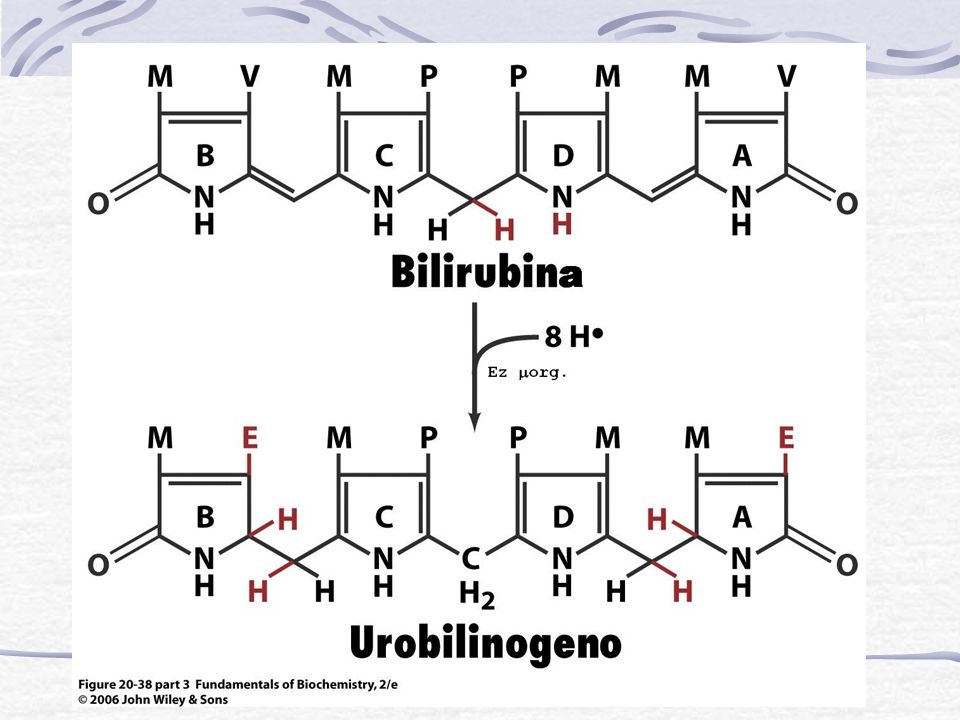
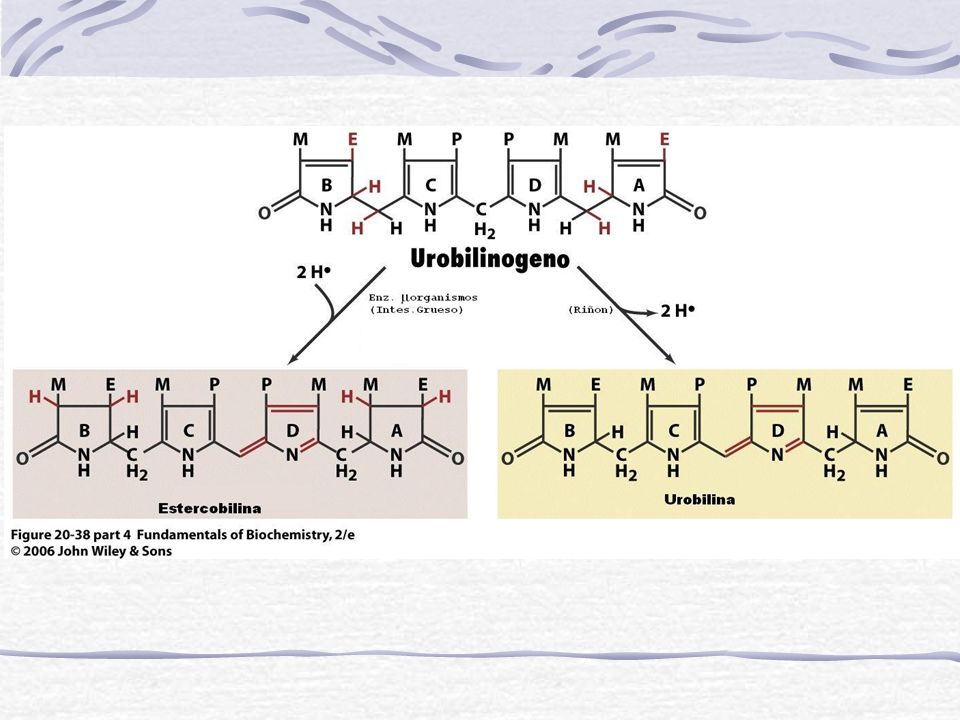
Bilirrubina conjugada La mayor parte se elimina por vía biliar hacia el duodeno Intestino Pigmentos fecales Bilirrubina es convertida en Urobilinogeno (incoloro) y Urobilina (marrón). Se elimina por las heces. 10-15% es reabsorvido y re-excretado a la bilis y otra parte por el riñón a la orina Una escasa cantidad de los conjugados se absorben desde el conducto biliar hacia el sistema sanguíneo y se elimina por vía renal
 y Urobilina (marrón). Se elimina por las heces % es reabsorvido y re-excretado a la bilis y otra parte por el riñón a la orina. Una escasa cantidad de los. conjugados se absorben desde el conducto biliar hacia el sistema sanguíneo y se elimina por vía renal.")
9
Transporte en plasma Albúmina RN: 0,5-1 mol B/ mol albúmina

11
Gen UDP-Glucuronosil-Transferasa 1- A1 (UGT1-A1)
1º Exón Variable (Especifico de Sustrato) Exones Comunes
 Exones Comunes.")
12
UGT1 A1 (RNAm UGT Bilirrubina)
locus UGT1 A Transcripto 1 Transcripto 6 UGT1 A (RNAm UGT- Fenol) 1A
 1A")
13
UDP-N-AcG (+) 2UDP-Acido glucurónico UDP-glucuronil transferasa (Gt)
2U+ 2PPi NDPasa

15
Bile salt export pump (BSEP) [o as sister of p-glycoprotein (SPGP)]:
Multidrug resistance- related protein (MRP2) [o canalicular multispecific organic anion transporter (cMOAT)]: transporta la mayoría de aniones que no sean ácidos biliares, incluyendo bilirrubina conjugada. Bile salt export pump (BSEP) [o as sister of p-glycoprotein (SPGP)]: Es el mayor transportador de ácidos biliares. MDR, multi drug resistance 3: Transporta FL desde la cara interna a la externa (hacia el canalículo biliar) Familial Intrahepatic Cholestasis1: transloca FL (como fosfatidilserina y fosfatidiletanolamina)
![Bile salt export pump (BSEP) [o as sister of p-glycoprotein (SPGP)]:](http://slideplayer.es/slide/138605/2/images/15/Bile+salt+export+pump+%28BSEP%29+%5Bo+as+sister+of+p-glycoprotein+%28SPGP%29%5D%3A.jpg "Multidrug resistance- related protein (MRP2) [o canalicular multispecific organic anion transporter (cMOAT)]: transporta la mayoría de aniones que no sean ácidos biliares, incluyendo bilirrubina conjugada. Bile salt export pump (BSEP) [o. as sister of p-glycoprotein (SPGP)]: Es el mayor transportador de ácidos biliares. MDR, multi drug resistance 3: Transporta FL desde la cara interna a la externa (hacia el canalículo biliar) Familial Intrahepatic Cholestasis1: transloca FL (como fosfatidilserina y fosfatidiletanolamina)")
17
= MDR3 = BSEP

18
cMOAT=MDR2=ABC C2 6 DTM 5 DTM 6 DTM

19
HIPERBILIRRUBINEMIAS

20
Historia - La Ictericia Neonatal se ha reconocido por centurias
1875: Primera descripción anatómica de cerebros ictéricos por Johannes Orth. 1904: El termino Kernicterus fue acuñado por Christian Georg Schmorl después de la examinación postmortem de 120 cerebros ictéricos de infantes. : Aumenta el conocimiento del metabolismo de bilirubina. 1958: Fototerapia para ictericia. Hna Jean Ward, Dr. Cremer, y Jerry Lucey (1968).
.")
21
Determinación de Bilirrubina
530 nm RN Termino Prematuros Hasta 24 hs 6 mg/dl 8 mg/dl Hasta las 48 hs 7,5 mg/dl 12 mg/dl Del 3-5° día 24 mg/dl Adulto: 1,0 mg/dl ---

22
Determinación de Bilirrubina
530 nm: azo-bilirrubina Valores Normales en Adultos: Bilirrubina Conjugada (Directa): hasta 0,2 mg/dl Bilirrubina No Conjugada (Indirecta) 0,8 mg/dl Total: hasta 1mg/dl
: hasta 0,2 mg/dl. Bilirrubina No Conjugada (Indirecta) 0,8 mg/dl. Total: hasta 1mg/dl.")
23
ICTERICIA Coloración amarillenta de la piel, mucosas y conjuntivas causadas por cifras de bilirrubina en sangre superiores a las normales Cuando la Bilirrubina Total > 2,5 mg/dl los tejidos toman el color de la Bilirrubina.

24
ICTERICIAS DEL RECIÉN NACIDO
Ictericia: alteración clínica mas frecuente en el periodo neonatal Todos los RN presentan BilirrubinaTotal > 2mg/dl en la 1ª semana de vida 25-50 % supera los 7 mg/dl < 10 % RN ictericia patológica

25
ICTERICIA FISIOLÓGICA
Aumento de bilirrubina no conjugada Máximo de 6-8 mg/dl al 3º día de vida Desaparición de la ictericia clínica a los 7-10 días de vida

26
CRITERIOS DE ICTERICIA NO FISIOLÓGICA
Una ictericia no es fisiológica si: a- Aparece en la primeras 24 horas de vida (hemólisis) b- Supera el valor máximo de 13 mg/dl (15 si está amamantado con leche materna) c- Persistencia de la ictericia durante mas de semanas e- Incremento en la bilirrubinemia > 5mg/dl por día
 b- Supera el valor máximo de 13 mg/dl (15 si está amamantado con leche materna) c- Persistencia de la ictericia durante mas de 2 semanas. e- Incremento en la bilirrubinemia > 5mg/dl por día.")
27
SIGNOS DE ALARMA EN EL R.N. ICTÉRICO
Historia familiar de enfermedad hemolítica Vómitos Letargia Rechazo del alimento Fiebre o hipotermia Inicio de la ictericia en el primer día o prolongación después de diez días Coluria Acolia

28
TOXICIDAD DE LA BILIRRUBINA
Bilirrubina. Sustancia lipofílica, insoluble en agua. Necesita ser vehiculizada por la Albúmina. Descopla la fosforilación oxidativa en las mitocondrias cerebrales. Inhibe la actividad de ATPasa de las mitocondrias del cerebro Induce la inflamación en el cerebro NEUROTOXICIDAD “KERNICTERUS”

29
Ictericia Nuclear de Schmorl o Kernicterus: necrosis de los núcleos extrapiramidales de la base del encéfalo por impregnación de Bilirrubina sin conjugar

30
CLÍNICA DEL KERNICTERUS
Se observan 3 Fases: (1) Hipotonía, letargia, llanto agudo, mala succión. (2) Hipertonía musculatura extensora (opistotonos), fiebre, convulsiones. (3) Hipotonía Secuelas: Diplejía espática. Encefalopatía crónica: Atetosis, sordera neurosensorial
 Hipotonía, letargia, llanto agudo, mala succión. (2) Hipertonía musculatura extensora (opistotonos), fiebre, convulsiones. (3) Hipotonía. Secuelas: Diplejía espática. Encefalopatía crónica: Atetosis, sordera neurosensorial.")
31
Médula N. Estriado, Hipocampo

32
Kernicterus:

33
HIPERBILIRRUBINEMIAS NO HEMOLITICAS: CAUSAS
1. Aumento de la producción de bilirrubina 2. Disminución de la captación y/o conjugación hepática 3. Alteración de la excreción biliar

34
Clasificación: Hiperbilirrubinemias No Conjugadas Síndrome de Gilbert
Síndrome de Crigler-Najjar, tipo I y II Hiperbilirrubinemias Conjugadas Síndrome de Dubin-Johnson Síndrome de Rotor

36
Síndrome de Gilbert El síndrome de Gilbert es clásicamente definido como una hiperbilirrubinemia intermitente crónica, suave sin signos manifiestos de hemólisis. Esta condición esta asociada con una variedad de síntomas, tales como fatiga, astenia, dispepsia y letargo. Clínicamente su comienzo ocurre en la pubertad y consiste en manifestaciones de ictericia intermitente con variables elevaciones de bilirrubina no conjugada de 1-4 mg/dl

37
Síndrome de Gilbert Laboratorio:
Hepatograma, (transaminasas (GOT/GPT), FAL, -GTP): normales Curvas de depuración anormales
, FAL, -GTP): normales. Curvas de depuración anormales.")
38
Bromosulfoftaleína (BSP)
Verde de indocianina (ICG)
")
39
1º caída Bilirrubina: 3 caídas BSP: 2 caídas ICG: 1 caída

40
S. Gilbert

41
Síndrome de Gilbert Laboratorio: Curvas de depuración anormales:
Algunos pacientes poseían anormal el primer exponencial indicando un problema en la captación (quizás en la permeasa?) Otros encontraban anormal el segundo exponencial reflejando esto una deficiente biotransformación (baja actividad de la UDPGt ?) y Otros autores hallaban que tanto el primer como el segundo exponencial eran anormales
 Otros encontraban anormal el segundo exponencial reflejando esto una deficiente biotransformación (baja actividad de la UDPGt ) y. Otros autores hallaban que tanto el primer como el segundo exponencial eran anormales.")
42
Síndrome de Gilbert Defecto Molecular:
Alelo A(TA)7 TAA (inserción TA); 30% de la actividad de UGT1-A1
7 TAA (inserción TA); 30% de la actividad de UGT1-A1.")
43
Síndrome de Gilbert Laboratorio:
Test de ácido nicotínico (sobrecarga de B)
")
44
Síndrome de Crigler-Najjar
El defecto metabólico es la ausencia de actividad en hígado de Bilirrubina-Uridin Difosfatoglucuronosil transferasa (UDP-Gt). Se hereda AR Hay 2 tipos: Tipo I: severa y Tipo II con 10% actividad
. Se hereda AR. Hay 2 tipos: Tipo I: severa y. Tipo II con 10% actividad.")
45
Síndrome de Crigler-Najjar
Diagnóstico Diferencial de los Síndromes de Crigler-Najjar TIPO I TIPO II Actividad de UDP-Gt No detectable Marcadamente disminuida Bilirrubina en Bilis No Conjugada y B(AG) (trazas) B(AG)2 (trazas) Efectos de Fenobarbital sobre Bilirr. sérica No tiene efectos Importante Efecto de la mutación Ausencia de actividad 10% actividad
 (trazas) B(AG)2 (trazas) Efectos de Fenobarbital sobre Bilirr. sérica. No tiene efectos. Importante Efecto de la mutación. Ausencia de actividad. 10% actividad.")
46
Síndrome de Crigler-Najjar Características Clínicas
Tipo I Severa ictericia que aparece entre el 1er y 3er día después del nacimiento La [bilirrubina]= mg /dl Hay kernicterus o encefalopatía por bilirrubina Tipo II - La [bilirrubina]= mg/dl - kernicterus: raro - Presentan una mayor respuesta a drogas que estimulan la hiperplasia del RE por ejemplo fenobarbital o dilantina

47
Síndrome de Crigler-Najjar Características Clínicas
200mg/día por 2 días Tratamiento para el tipo II

48
Síndrome de Crigler-Najjar Biología Molecular

49
Síndrome de Crigler-Najjar Biología Molecular

50
Síndrome de Crigler-Najjar
Laboratorio: Los niños con síndrome de Crigler-Najjar con bilirrubina sérica mayor de 20 mg/dl se consideran clínicamente como pacientes de alto riesgo Descartar otras causas de hiperbilirrubinemias, durante el rastreo neonatal

51
Síndrome de Crigler-Najjar
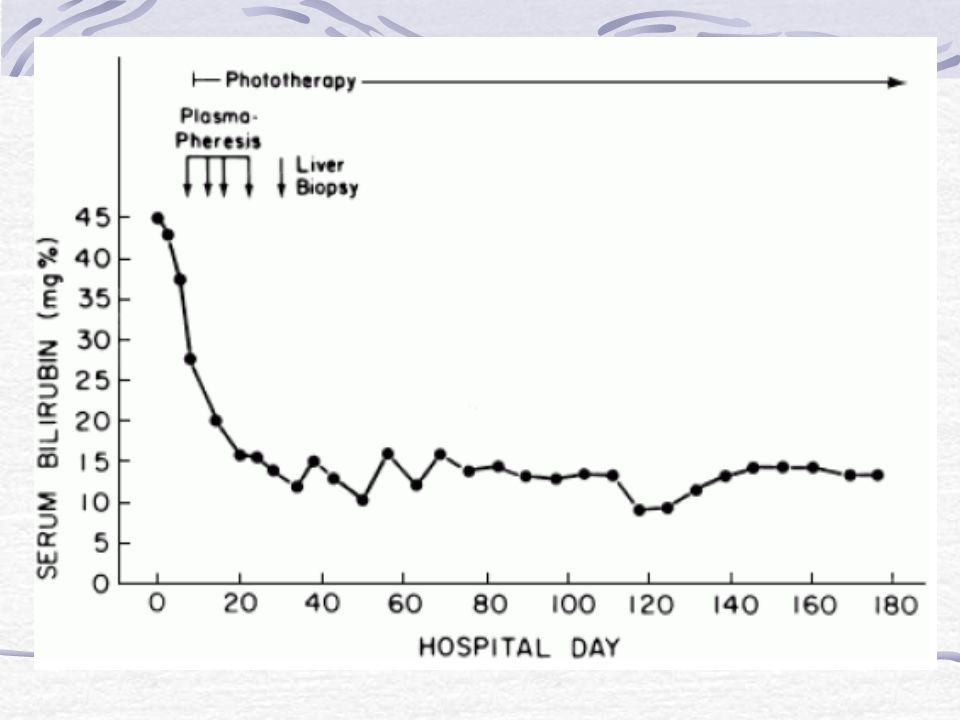
Tratamiento para el tipo I: Transplante de hígado. Plasmaféresis. Exanguinotransfusión. Fototerapia en el recién nacido. Sn protoporfirina IX (en etapa experimental)
")
52
TRATAMIENTO DE UNA ICTERICIA NEONATAL
Eliminar la bilirrubina no conjugada: Fototerapia En 1950 la Hna Ward del Hospital General de Rochford en Essex, notó que la luz diurna, desvanecía la coloración amarillenta de la piel de los recién nacidos

53
FOTOTERAPIA Espectro de Luz: 480-500 nm
Tubos fluorescentes azul (especial): 10 cm del bebe
: 10 cm del bebe.")
54
Z y E, proviene del aleman zusammen (juntos) y entgegen (opuestos), y son prefijos usados para designar la estereoquímica alrededor de un doble unión.
 y entgegen (opuestos), y son prefijos usados para designar la estereoquímica alrededor de un doble unión.")
56
INDICACIONES DE LA FOTOTERAPIA

58
Plasmaféresis El procedimiento básico consiste en la extracción de sangre, separación de las células sanguíneas del plasma, y la devolución de estas células de la sangre a la circulación del cuerpo, diluido con plasma fresco o en un sustituto. El sustituto más común es una solución salina esterilizada con proteína de albúmina humana. Durante el curso de una misma sesión, dos a tres litros de plasma se retira y se sustituye. La plasmaferesis es el método más eficiente para reducir la concentración de bilirrubina sérica durante la crisis de los pacientes con C-N tipo I

60
Clasificación: Hiperbilirrubinemias No Conjugadas Síndrome de Gilbert
Síndrome de Crigler-Najjar, tipo I y II Hiperbilirrubinemias Conjugadas Síndrome de Dubin-Johnson Síndrome de Rotor

61
Síndrome de Dubin-Johnson
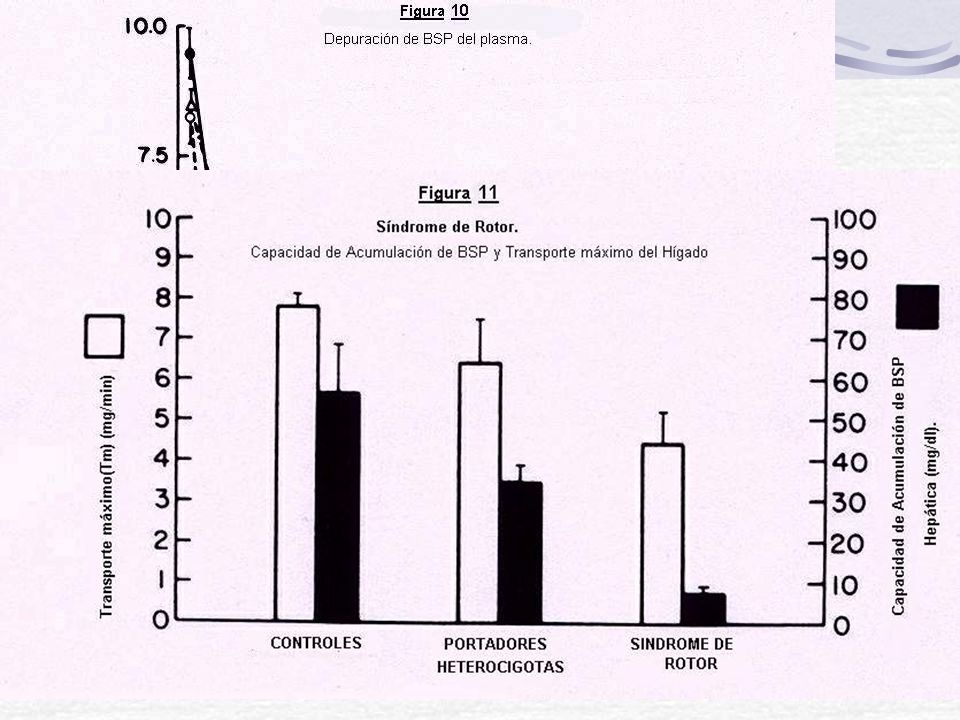
Herencia: AR, con una alta prevalencia estimada en un mínimo de 0,07 % (1/1400) y está frecuentemente asociado (60%) con deficiencia del factor VII de coagulación. Bilirrubina= 2-6 mg /dl (BC) Defecto: MRP2=cMOAT= ABC C2 Colecistografía Oral: uso de ácido Iopanoico (Colesom), tiene la particularidad de ser excretado por el hígado hacia las vías biliares y acumulación en la vesícula. Es (-) para los pacientes con D-J. Hay acumulación de Pigmentos en Hígado Curva de BSP anormal: min hay regurgitación
 y está frecuentemente asociado (60%) con deficiencia del factor VII de coagulación. Bilirrubina= 2-6 mg /dl (BC) Defecto: MRP2=cMOAT= ABC C2. Colecistografía Oral: uso de ácido Iopanoico (Colesom), tiene la particularidad de ser excretado por el hígado hacia las vías biliares y acumulación en la vesícula. Es (-) para los pacientes con D-J. Hay acumulación de Pigmentos en Hígado. Curva de BSP anormal: min hay regurgitación.")
62
Síndrome de Dubin-Johnson
Curva de BSP anormal: min hay regurgitación - Tm - Capacidad de retención hepatica: N

63
Síndrome de Dubin-Johnson
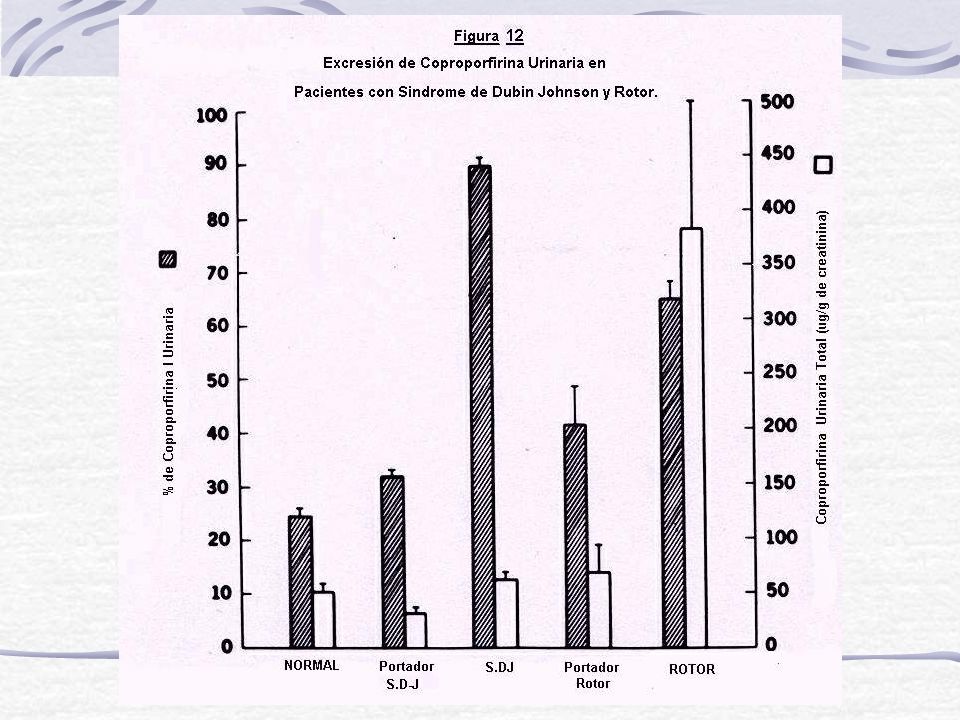
Excreción Urinarias de Copro Anormal: Solo D-J altera el patrón de excreción siendo 80% de esas Copro de tipo I TRATAMIENTO: No posee terapias, ya que es de evolución benigna.

64
Síndrome de Dubin-Johnson
Biología Molecular:

65
Síndrome de Rotor Herencia: AR Defecto:
El nombre oficial del gen es SLCO1 B1 (“solute carrier organic anion transporter family, member 1B1.”). Ha tenido otros nombres: LIVER-SPECIFIC TRANSPORTER 1 (LST1); ORGANIC ANION TRANSPORTER 2, (OATP2) ORGANIC ANION TRANSPORTER C, (OATPC) ORGANIC ANION TRANSPORTER 1B1, (OATP1B1) Función: La proteína codificada es un receptor transmembrana que media la captación de (independiente de sodio) de numerosos compuestos endógenos incluyendo bilirrubina, 17-beta- glucuronosil estradiol, taurocolato, metotrexato, sulfato de dehidroepiandrosterona, sulfato de estrona, la prostaglandina E2, el tromboxano B2, leucotrieno C3, leucotrienos E4, T4 y T3. Esta proteína también está implicada en la eliminación de fármacos complejos tales como las estatinas, bromosulfoftaleína y rifampicina de la sangre hacia los hepatocitos.
. Ha tenido otros nombres: LIVER-SPECIFIC TRANSPORTER 1 (LST1); ORGANIC ANION TRANSPORTER 2, (OATP2) ORGANIC ANION TRANSPORTER C, (OATPC) ORGANIC ANION TRANSPORTER 1B1, (OATP1B1) Función: La proteína codificada es un receptor transmembrana que media la captación de (independiente de sodio) de numerosos compuestos endógenos incluyendo bilirrubina, 17-beta- glucuronosil estradiol, taurocolato, metotrexato, sulfato de dehidroepiandrosterona, sulfato de estrona, la prostaglandina E2, el tromboxano B2, leucotrieno C3, leucotrienos E4, T4 y T3. Esta proteína también está implicada en la eliminación de fármacos complejos tales como las estatinas, bromosulfoftaleína y rifampicina de la sangre hacia los hepatocitos.")
68
Síndrome de Rotor: Diferencias D-J
Síndrome de Dubin-Johnson Síndrome de Rotor Gen afectado MRP2 (cMOAT) SLCO1 B1 Ictericia Intensidad Extensión en tiempo - Moderada, ocasionalmente, picos de 20 mg/dl - Puede fluctuar con estrógenos o embarazo - Moderada, ocasionalmente alta. - Fluctuante Hígado Inspección Microscópica Histología - Hígado Negro - Gránulos de pigmentos en hepatocitos centro -lobulares - Color pardo Normal. - Normal Colecistografía Cavidad biliar NO visualizada Normal Pronostico Benigno
 SLCO1 B1. Ictericia. Intensidad. Extensión en tiempo. - Moderada, ocasionalmente, picos de 20 mg/dl. - Puede fluctuar con estrógenos o embarazo. - Moderada, ocasionalmente alta. - Fluctuante. Hígado. Inspección Microscópica. Histología. - Hígado Negro. - Gránulos de pigmentos en hepatocitos centro -lobulares. - Color pardo Normal. - Normal. Colecistografía. Cavidad biliar NO visualizada. Normal. Pronostico. Benigno.")
69
RESUMEN GENERAL

70
S. Crigler-Najjar Tipo I S. Crigler-Najjar Tipo II
HIPERBILIRRUBINEMIA Historia, Examinación Física, Hepatograma Completo y Ácidos Biliares Séricos Totales. Recuento Celulares y de Reticulocitos en Sangre Periférica. Anormal Normal Sospecha de daño hepático Sospecha de Hiperbilirrubinemia Familiar No Conjugada (Indirecta) Conjugada (Directa) Exclusión de manifestaciones de hemólisis Bilirrubina Sérica (mg/dl Bilirrubina Sérica (mg/dl 2-6 Depuración de BSP e ICG < S. Dubin-Johnson S. de Gilbert S. Crigler-Najjar Tipo I S. Crigler-Najjar Tipo II S. Rotor
 Conjugada (Directa) Exclusión de manifestaciones de hemólisis. Bilirrubina Sérica (mg/dl. Bilirrubina Sérica (mg/dl Depuración de BSP e ICG. < 5 S. Dubin-Johnson. S. de Gilbert. S. Crigler-Najjar Tipo I. S. Crigler-Najjar Tipo II. S. Rotor.")
71
Síndrome de Crigler-Najjar
Característica Síndrome de Gilbert Síndrome de Crigler-Najjar Tipo I Tipo II Concentración Bilirrubina sérica (mg/dl) 1-5 25 6-25 Activ. Bilirrubina-UDP-glucuronil transferasa 60% 0% 10% Depuración de BSP Anormal 1/3 de los casos No Depuración de ICG Anormal 1/5 de los casos Respuesta al Fenobarbital Si Kernicterus Rara Herencia AR
 1-5. Activ. Bilirrubina-UDP-glucuronil transferasa. 60% 0% 10% Depuración de BSP Anormal. 1/3 de los casos. No. Depuración de ICG Anormal. 1/5 de los casos. Respuesta al Fenobarbital. Si. Kernicterus. Rara. Herencia. AR.")
72
Concentrac. Bilirrubina sérica (mg/dl) 2-6
Característica Sínd. de Dubin-Johnson Sindrome de Rotor Concentrac. Bilirrubina sérica (mg/dl) 2-6 Actividad Bilirrubina-UDP-glucuronil transferasa Normal Depuración de BSP Anormal Si (regurgitación) a los 40’ Si (pronunciado) Depuración de ICG Anormal No Si Colecistografía Oral de la Cavidad Vesicular Usualmente NO visualizable Usualmente visualizable Acumulación de pigmentos en hígado Coproporfirinas Aumentadas (I 80% del total) Aumento de isómeros I y III (I 80% del total) Sitio del déficit Secreción canalicular(MRP2) Transportador de Bilirrubina (SLCO1 B1 )
 2-6. Actividad Bilirrubina-UDP-glucuronil transferasa. Normal. Depuración de BSP Anormal. Si (regurgitación) a los 40’ Si (pronunciado) Depuración de ICG Anormal. No. Si. Colecistografía Oral de la Cavidad Vesicular. Usualmente NO visualizable. Usualmente visualizable. Acumulación de pigmentos en hígado. Coproporfirinas. Aumentadas (I 80% del total) Aumento de isómeros I y III (I 80% del total) Sitio del déficit. Secreción canalicular(MRP2) Transportador de Bilirrubina (SLCO1 B1 )")
73
Bibliografia: Lajos Okolocsanyi, Gino Nassuato and Mario Strazzabosco: Familial Hyperbilirubinemias. Clinical Aspects. Chapter 44. Hepatic transport and Bile secretion: Physiology and Pathophysiology. Raven Press, Ltd., New York Gerard Odell and Glenn Gourley: Hereditary hyperbilirubinemia. Chapter 67. Texbook of Gastroenterology and Nutrition in Infancy. Edited by E. Lebenthal. Raven Press, Ltd., New York. 1989 Donald Ostrow, Pesupati Mujerjee and Claudio Tiribelli: Structure and binding of unconjugated bilirubin: relevance for physiological and pathophysiological function. Review. J. Lipid Res 1994, 35; Charles R. Scriver, William S. Sly, Barton Childs, Arthur Beaudet, William Sly, David Valle, Kenneth W. Kinzler PhD, Bert Vogelstein MD The Metabolic and Molecular Bases of Inherited Disease, 4 volume set New York, McGraw Hill; pag van den Bergh AAH, Muller P: Ueber eine direkte und eine indirekte Diazoreaktion auf Bilirubin. Biochem Z. 77:90, 1916. Schmorl G: Zur Kenntnis des ikterus neonatatorum, inbesondere der dabei auftreten den gehirnveranderungen. Verh Dtsch Ges Pathol. 6:109, 1903. Jayanta Roy Chowdhury, Allan W. Wolkoff, Namita Roy Chowdhury, Irwin M. Arias. PART 13: PORPHYRINS- Chapter 125: Hereditary Jaundice and Disorders of Bilirubin Metabolism. En Scriver CR, Beaudet AL, Sly W S, Valle D (eds). The online metabolic & molecular bases of inherited disease. New York, McGraw Hill, 2012; pag Giovanni Agati and Franco Fusi: New Trends in photobiology. Recents advances in bilirubin photophysics. J. Photochem. Photobiol 1990, 7: 1-14. PA Gustafson and DW Boyle: Bilirubin Index. Medical Hypotheses 1995, 45: A Robertson, W Karp and R Brodersen: Bilirubin displacing effect of drugs in neonatology. Acta Paediatr Scand 1991: 80: Burchell A, Hume R Molecular genetics basis of Gilbert’s syndrome. J Gastroenterol. Hepatol. 14, Sampietro M, Lupica L, Pettero L, Romano R, Molteni V, Fionelli G TATA-box promoter mutant in the promotor of UDP-glucuronyltransferase gene in Italian patients with Gilbert’s síndrome. Ital J Gastroenterol Hepatol. 30: Beutler E, Gelbert T, Demina A Racial in the UDP-glucuronyltransferase 1 promoter: a balanced polymorfism for regulation of bilirrubin metabolism. PNAS, USA 95: Tukey RH, Strassburg CP Human UDP-Glucuronosyl-transferases: Metabolism, Expression and Disease. Annu. Rev. Pharmacol. Toxicol. 40, Namita Roy Chowdhury, Irwin M. Arias, Allan W. Wolkoff and Jayanta Roy Chowdhury The Liver-Biology and Pathobiology, Fourth Edition- Chapter 20. DISORDERS OF BILIRUBIN
. The online metabolic & molecular bases of inherited disease. New York, McGraw Hill, 2012; pag Giovanni Agati and Franco Fusi: New Trends in photobiology. Recents advances in bilirubin photophysics. J. Photochem. Photobiol 1990, 7: PA Gustafson and DW Boyle: Bilirubin Index. Medical Hypotheses 1995, 45: A Robertson, W Karp and R Brodersen: Bilirubin displacing effect of drugs in neonatology. Acta Paediatr Scand 1991: 80: Burchell A, Hume R Molecular genetics basis of Gilbert’s syndrome. J Gastroenterol. Hepatol. 14, Sampietro M, Lupica L, Pettero L, Romano R, Molteni V, Fionelli G TATA-box promoter mutant in the promotor of UDP-glucuronyltransferase gene in Italian patients with Gilbert’s síndrome. Ital J Gastroenterol Hepatol. 30: Beutler E, Gelbert T, Demina A Racial in the UDP-glucuronyltransferase 1 promoter: a balanced polymorfism for regulation of bilirrubin metabolism. PNAS, USA 95: Tukey RH, Strassburg CP Human UDP-Glucuronosyl-transferases: Metabolism, Expression and Disease. Annu. Rev. Pharmacol. Toxicol. 40, Namita Roy Chowdhury, Irwin M. Arias, Allan W. Wolkoff and Jayanta Roy Chowdhury The Liver-Biology and Pathobiology, Fourth Edition- Chapter 20. DISORDERS OF BILIRUBIN.")
74
Alguna pregunta?? Muchas gracias por su atención!

Presentaciones similares