Descargar la presentación
La descarga está en progreso. Por favor, espere

1
Patología General. Introducción a la Medicina Clínica
Remigio Cordero Torres Badajoz 10 de Septiembre de 2015

2
William Osler (Band Head, 1849 - Oxford, 1919)
")
3
“Arrancar a la naturaleza los secretos que han dejado perplejos a los filósofos de todos los tiempos, seguir el rastro hasta su origen a las causas de la enfermedad, correlacionar los inmensos almacenes de conocimientos para que puedan estar fácilmente disponibles para la prevención y cura de la enfermedad. Esas son nuestras ambiciones” William Osler.Chauvinismo en Medicina1902.

4
“Arrancar a la naturaleza los secretos que han dejado perplejos a los filósofos de todos los tiempos, seguir el rastro hasta su origen a las causas de la enfermedad, correlacionar los inmensos almacenes de conocimientos para que puedan estar fácilmente disponibles para la prevención y cura de la enfermedad. Esas son nuestras ambiciones” William Osler.Chauvinismo en Medicina1902.

5
PATOLOGÍA: Estudio de la enfermedad
ETIOLOGÍA: causa de la enfermedad PATOGENIA: mecanismos por los que los agentes causales ocasionan las lesiones FISIOPATOLOGÍA: estudio trastornos que se producen en la función y estructura de órganos y sistemas. De estos trastornos derivan las manifestaciones clínicas.

6
Agente Causal o Etiológico PATOGENIA
Alteraciones funcionales- estructurales FISIOPATOLOGÍA Manifestaciones Clínicas Síntomas y Signos: SÍNDROMES

7
FENOTIPO = GENOTIPO + AMBIENTE
Resultado de la constitución genetica heredada más los factores ambientales Enfermedad: Resultado de la interacción entre la constitución genética y el entorno

8
La genómica modifica aspectos básicos de la medicina
Naturaleza del contrato médico Perspectiva de la enfermedad: PREPACIENTES Contexto social y cultural: discriminación potencial.

9
PREPACIENTES Nancy Wexler sostiene en brazos a un niño con un caso precoz de la enfermedad de Huntington, Lago Maracaibo. Venezuela

11
The Life Cycle of Human Immunodeficiency Virus Type 1 (HIV-1), Showing Potential Targets for Antiretroviral Therapy Figure 1. The Life Cycle of Human Immunodeficiency Virus Type 1 (HIV-1), Showing Potential Targets for Antiretroviral Therapy. HIV-1 binds to receptors on the cell surface, undergoes membrane fusion, and then releases copies of the RNA genome into the cytoplasm. After successful invasion of the cell, the viral reverse-transcriptase enzyme transcribes single-stranded viral RNA into double-stranded DNA that can be integrated into the genetic material of the human host. Reverse-transcriptase inhibitors were the first agents approved for the treatment of HIV-1; currently available inhibitors of this enzyme are nucleoside antagonists (zidovudine, didanosine, zalcitabine, lamivudine, stavudine, abacavir, and combined formulations), nonnucleoside competitive inhibitors (nevirapine, delavirdine, and efavirenz), and one nucleotide analogue (tenofovir). The viral integrase enzyme is required for the integration of proviral DNA into the host genome before replication. Investigational integrase inhibitors are currently in early clinical trials. When the infected cell synthesizes new protein, integrated proviral DNA is also translated into the protein building blocks of new viral progeny. The viral components then assemble on the cell surface and bud out as immature viral particles. The final maturation of newly formed viruses requires the HIV-1 protease to digest larger components into the intricate pieces that make up an infectious virion. Several protease inhibitors (ritonavir, indinavir, nelfinavir, amprenavir, lopinavir-ritonavir, and two formulations of saquinavir) are currently in clinical use. Kilby J and Eron J. N Engl J Med 2003;348:
, Showing Potential Targets for Antiretroviral Therapy. HIV-1 binds to receptors on the cell surface, undergoes membrane fusion, and then releases copies of the RNA genome into the cytoplasm. After successful invasion of the cell, the viral reverse-transcriptase enzyme transcribes single-stranded viral RNA into double-stranded DNA that can be integrated into the genetic material of the human host. Reverse-transcriptase inhibitors were the first agents approved for the treatment of HIV-1; currently available inhibitors of this enzyme are nucleoside antagonists (zidovudine, didanosine, zalcitabine, lamivudine, stavudine, abacavir, and combined formulations), nonnucleoside competitive inhibitors (nevirapine, delavirdine, and efavirenz), and one nucleotide analogue (tenofovir). The viral integrase enzyme is required for the integration of proviral DNA into the host genome before replication. Investigational integrase inhibitors are currently in early clinical trials. When the infected cell synthesizes new protein, integrated proviral DNA is also translated into the protein building blocks of new viral progeny. The viral components then assemble on the cell surface and bud out as immature viral particles. The final maturation of newly formed viruses requires the HIV-1 protease to digest larger components into the intricate pieces that make up an infectious virion. Several protease inhibitors (ritonavir, indinavir, nelfinavir, amprenavir, lopinavir-ritonavir, and two formulations of saquinavir) are currently in clinical use. Kilby J and Eron J. N Engl J Med 2003;348:")
12
Agente Causal o Etiológico PATOGENIA
Alteraciones funcionales- estructurales FISIOPATOLOGÍA Manifestaciones Clínicas Síntomas y Signos: SÍNDROMES

13
“Arrancar a la naturaleza los secretos que han dejado perplejos a los filósofos de todos los tiempos, seguir el rastro hasta su origen a las causas de la enfermedad, correlacionar los inmensos almacenes de conocimientos para que puedan estar fácilmente disponibles para la prevención y cura de la enfermedad. Esas son nuestras ambiciones” William Osler.Chauvinismo en Medicina1902.

14
La patología y la clínica representan los aspectos teóricos y práctico de la medicina clínica.
La actividad clínica, aplica unos conocimientos científicos – los de la patología- con un método científico y utiliza los recursos de una técnica cada vez más refinada y compleja.

15
MEDICINA CLÍNICA DIAGNÓSTICO PRONÓSTICO TRATAMIENTO

16
CLÍNICA Actividad que realiza el médico junto al enfermo
- DIAGNÓSTICO Recogida de Datos Análisis e interpretación de los datos Identificación de Síndrome Diagnóstico diferencial Identificación etiología - PRONÓSTICO - TRATAMIENTO

17
Agente Causal o Etiológico PATOGENIA
CLINICA Alteraciones funcionales- estructurales FISIOPATOLOGÍA Manifestaciones Clínicas Síntomas y Signos: SÍNDROMES

18
CLÍNICA Actividad que realiza el médico junto al enfermo
- DIAGNÓSTICO Recogida de Datos Análisis e interpretación de los datos Identificación de Síndrome Diagnóstico diferencial Identificación etiología - PRONÓSTICO - TRATAMIENTO

19
Razonamiento Clínico Conocimientos biomédicos
Conocimientos Fisiopatológicos Conocimientos adaptados a práctica clínica

20
“Arrancar a la naturaleza los secretos que han dejado perplejos a los filósofos de todos los tiempos, seguir el rastro hasta su origen a las causas de la enfermedad, correlacionar los inmensos almacenes de conocimientos para que puedan estar fácilmente disponibles para la prevención y cura de la enfermedad. Esas son nuestras ambiciones” William Osler.Chauvinismo en Medicina1902.

21
1997 : Wilmunt & Campbell; Instituto Roslin

25
Progresos año 2000 Secuenciación genómica (Science)
Secuenciación genoma humano (AHA) “Libro de la vida”, “Santo Grial de la biología” “ Entrar en una nueva era”
 Libro de la vida , Santo Grial de la biología Entrar en una nueva era")
26
Hitos posteriores 2002. Síntesis de un virus en un tubo de ensayo (Eckard Wimmer) 2003. Síntesis de un bacteriófago (JCraig Venter) 2007. Síntesis de ADN bacteria Mycoplasma genitalium (Daniel Gibson) 2010. Creación de una nueva especie de laboratorio Mycoplasma mycoides JVC-syn1.0 (Craig Venter). 2014. Creación de un cromosoma artificial e inserción en un ser vivo Saccharomyces cerevisiae (Jef Boeke) pares de bases añadidas.
 Creación de una nueva especie de laboratorio Mycoplasma mycoides JVC-syn1.0 (Craig Venter) Creación de un cromosoma artificial e inserción en un ser vivo Saccharomyces cerevisiae (Jef Boeke) pares de bases añadidas.")
29
Clonación reproductiva vs clonación terapéutica
CLONACIÓN REPRODUCTIVA: creación de un individuo genéticamente idéntico a otro. CLONACIÓN TERAPÉUTICA: creación de células madre genéticamente idénticas a las células de un paciente, con el objetivo de utilizarlas para poder tratar o curar una enfermedad sin que se produzca ningún rechazo.

30
Paso 1 Masa Celular Interna

31
Paso 4

32
Paso 5 Cerebro Vasos Sanguíneos Pulmones Corazón Páncreas Hígado Riñón
Cartílago Hueso Músculo

34
Paso 5 Grupos células insulares

35
Células madres: Medicina Regenerativa
Autotransplantes de Médula Ósea Enfermedades degenerativas: Parkinson Alzheimer Enfermedades autoinmunes: diabetes Lesiones medulares Enfermedades cardiovasculares Cáncer Enfermedad de Crohn Regeneración piel y de pelo

36
Paso 1 Masa Celular Interna

37
Gearhart J et al. N Engl J Med 2007;357:1469-1472.
Induction of Pluripotent Stem Cells through Retroviral Transduction. Gearhart J et al. N Engl J Med 2007;357: Induction of Pluripotent Stem Cells through Retroviral Transduction. Retrovirally encoded transcription factor genes were introduced into mouse embryonic and adult fibroblasts. After integration and expression of the transgenes, the fibroblasts were reprogrammed to pluripotency.

38
Mummery C. N Engl J Med 2011;364:2160-2162.
Genetic Effects of Reprogramming Cells for Pluripotency. Figure 1. Genetic Effects of Reprogramming Cells for Pluripotency. Genetic lesions arise during reprogramming of fibroblasts to pluripotency (thus generating human induced pluripotent stem [hiPS] cells) and during prolonged culture of both hiPS cells and human embryonic stem (hES) cells. Recent studies show that in addition to gross chromosomal changes that occur during prolonged culture of hiPS and hES cells (e.g., duplication of parts of chromosomes 12 and 20), gene copy-number variations and point mutations can be induced during the reprogramming of somatic cells into hiPS cells, resulting in many more DNA lesions (by up to a factor of 10) in hiPS cells than in the somatic cells from which they are derived.1– 5 During prolonged culture, the frequency with which these new mutations are detected decreases. Mummery C. N Engl J Med 2011;364:
 and during prolonged culture of both hiPS cells and human embryonic stem (hES) cells. Recent studies show that in addition to gross chromosomal changes that occur during prolonged culture of hiPS and hES cells (e.g., duplication of parts of chromosomes 12 and 20), gene copy-number variations and point mutations can be induced during the reprogramming of somatic cells into hiPS cells, resulting in many more DNA lesions (by up to a factor of 10) in hiPS cells than in the somatic cells from which they are derived.1– 5 During prolonged culture, the frequency with which these new mutations are detected decreases. Mummery C. N Engl J Med 2011;364:")
39
EL PAÍS, sábado 13 de Septiembre de 2014
Japón realiza el primer trasplante en humanos de células iPS Una mujer con degeneración macular recibe una retina cultivada en laboratorio Las células iPS de pluripotencia inducida, se obtienen de células de la piel del paciente y se transforman en cualquiera de los tejidos y tipos celulares, de tal forma que se evita el rechazo inmunológico. En 2012 se concedió el premio Nobel de Medicina al japonés Shinya Yamanaka por desarrollar el método para reprogramar células adultas

40
En 2012 se concedió el premio Nobel de Medicina al japonés Shinya Yamanaka por desarrollar el método para reprogramar células adultas.

41
Haruko Obokata – Yoshiki Sasai
Haruko Obokata – Yoshiki Sasai . Células Stap (stimulus-triggered acquisition pluripotency ) Artículo publicado en Nature en Enero 2014 Obokata dimite en Diciembre de 2014
 Artículo publicado en Nature en Enero Obokata dimite en Diciembre de")
42
Medicina Regenerativa
Células Madre Terapia Génica Inserción de un gen para sustituir o bloquear un gen defectuoso Ingeniería de tejidos Andamiajes poliméricos + células vivas Vejiga, Uretra, Traquea… Corazón , Pulmón

43
“Arrancar a la naturaleza los secretos que han dejado perplejos a los filósofos de todos los tiempos, seguir el rastro hasta su origen a las causas de la enfermedad, correlacionar los inmensos almacenes de conocimientos para que puedan estar fácilmente disponibles para la prevención y cura de la enfermedad. Esas son nuestras ambiciones” William Osler.Chauvinismo en Medicina1902.

44
Hitos médicos más importantes del milenio ABCNEWS
1 ANTIBIÓTICOS. Fleming 1920 2 VACUNACIÓN. Jenner 1796 3 RAYOS X. Roentgen 1920 4 ANESTESIA. Oxido Nitroso 1772 5 HÉLICE ADN. Watson y Crick 1953 6 TEORÍA GERMEN. Pasteur 1861 7 TRANSPLANTE DE ÓRGANOS 8 HIGIENE Y DISPONIBILIDAD DE AGUA 9 APARATO CIRCULATORIO WilliamHarvey 1682 10 MICROSCOPIO Antoni van Leuwenhoek 1683

45
Contribución del avance tecnológico a la salud global
GLOBAL BURDEN OF DISEASE .GBD (OMS. Harvard Schooll of Public Health. BM) Causas de muerte por enfermedad Incidencia factores de enfermedad Muertes atribuibles a distintos factores de riesgo.
 Causas de muerte por enfermedad. Incidencia factores de enfermedad. Muertes atribuibles a distintos factores de riesgo.")
46
Causas de muerte por enfermedades (millares) British Med Journal 1997
PAÍSES DESARROLLADOS E Cardiovasculares Cáncer Lesiones Infecc respiratorias Diabetes Infecc/parasitarias Maternas/perinatales Deficiencias nutrición PAÍSES EN DESARROLLO Infecc/Parasitarias E Cardiovasculares Lesiones Infec respiratorias Cáncer Maternas/perinatales 2.812 Deficiencias nutrición Diabetes

47
MUERTES ATRIBUIDAS A DISTINTOS FACTORES DE RIESGO (millares)
Desnutrición Tabaco Hipertensión Agua/Higiene Sedentarismo Profesionales Sexo no seguro Alcohol Contaminación aire

49
Baize S et al. N Engl J Med 2014. DOI: 10.1056/NEJMoa1404505
Transmission Chains in the Outbreak of Ebola Virus Disease in Guinea. Figure 2. Transmission Chains in the Outbreak of Ebola Virus Disease in Guinea. Shown are transmission chains in the Ebola virus disease outbreak involving laboratory-confirmed cases. The presumed means of transmission of Zaire ebolavirus (EBOV), as revealed by epidemiologic investigation, are indicated by solid arrows. Dashed arrows indicate that the epidemiologic links are not well established. Laboratory-confirmed cases (C) are indicated with red circles, and suspected cases (S) are indicated with the case number. The inset image is an electron microscopic scan of the Guinean strain of EBOV in blood obtained from a patient. A typical complete virus particle, with the ends marked by arrows, and two degraded particles (arrowheads) are shown (scale bar, 100 nm). Baize S et al. N Engl J Med DOI: /NEJMoa
, as revealed by epidemiologic investigation, are indicated by solid arrows. Dashed arrows indicate that the epidemiologic links are not well established. Laboratory-confirmed cases (C) are indicated with red circles, and suspected cases (S) are indicated with the case number. The inset image is an electron microscopic scan of the Guinean strain of EBOV in blood obtained from a patient. A typical complete virus particle, with the ends marked by arrows, and two degraded particles (arrowheads) are shown (scale bar, 100 nm). Baize S et al. N Engl J Med DOI: /NEJMoa")
50
Baize S et al. N Engl J Med 2014. DOI: 10.1056/NEJMoa1404505
Transmission Chains in the Outbreak of Ebola Virus Disease in Guinea. Figure 2. Transmission Chains in the Outbreak of Ebola Virus Disease in Guinea. Shown are transmission chains in the Ebola virus disease outbreak involving laboratory-confirmed cases. The presumed means of transmission of Zaire ebolavirus (EBOV), as revealed by epidemiologic investigation, are indicated by solid arrows. Dashed arrows indicate that the epidemiologic links are not well established. Laboratory-confirmed cases (C) are indicated with red circles, and suspected cases (S) are indicated with the case number. The inset image is an electron microscopic scan of the Guinean strain of EBOV in blood obtained from a patient. A typical complete virus particle, with the ends marked by arrows, and two degraded particles (arrowheads) are shown (scale bar, 100 nm). Baize S et al. N Engl J Med DOI: /NEJMoa
, as revealed by epidemiologic investigation, are indicated by solid arrows. Dashed arrows indicate that the epidemiologic links are not well established. Laboratory-confirmed cases (C) are indicated with red circles, and suspected cases (S) are indicated with the case number. The inset image is an electron microscopic scan of the Guinean strain of EBOV in blood obtained from a patient. A typical complete virus particle, with the ends marked by arrows, and two degraded particles (arrowheads) are shown (scale bar, 100 nm). Baize S et al. N Engl J Med DOI: /NEJMoa")
51
Baize S et al. N Engl J Med 2014. DOI: 10.1056/NEJMoa1404505
Transmission Chains in the Outbreak of Ebola Virus Disease in Guinea. Figure 2. Transmission Chains in the Outbreak of Ebola Virus Disease in Guinea. Shown are transmission chains in the Ebola virus disease outbreak involving laboratory-confirmed cases. The presumed means of transmission of Zaire ebolavirus (EBOV), as revealed by epidemiologic investigation, are indicated by solid arrows. Dashed arrows indicate that the epidemiologic links are not well established. Laboratory-confirmed cases (C) are indicated with red circles, and suspected cases (S) are indicated with the case number. The inset image is an electron microscopic scan of the Guinean strain of EBOV in blood obtained from a patient. A typical complete virus particle, with the ends marked by arrows, and two degraded particles (arrowheads) are shown (scale bar, 100 nm). Baize S et al. N Engl J Med DOI: /NEJMoa
, as revealed by epidemiologic investigation, are indicated by solid arrows. Dashed arrows indicate that the epidemiologic links are not well established. Laboratory-confirmed cases (C) are indicated with red circles, and suspected cases (S) are indicated with the case number. The inset image is an electron microscopic scan of the Guinean strain of EBOV in blood obtained from a patient. A typical complete virus particle, with the ends marked by arrows, and two degraded particles (arrowheads) are shown (scale bar, 100 nm). Baize S et al. N Engl J Med DOI: /NEJMoa")
52
Baize S et al. N Engl J Med 2014. DOI: 10.1056/NEJMoa1404505
Map of Guinea Showing Initial Locations of the Outbreak of Ebola Virus Disease. Figure 1. Map of Guinea Showing Initial Locations of the Outbreak of Ebola Virus Disease. The area of the outbreak is highlighted in red. The main road between the outbreak area and Conakry, the capital of Guinea, is also shown. The map was modified from a United Nations map. Baize S et al. N Engl J Med DOI: /NEJMoa

53
Briand S et al. N Engl J Med 2014. DOI: 10.1056/NEJMp1409858
Numbers of Confirmed and Probable Ebola Cases Reported Weekly from Guinea, Sierra Leone, and Liberia from December 23, 2013, to August 11, 2014. Leone, and Liberia from December 23, 2013 Numbers of Confirmed and Probable Ebola Cases Reported Weekly from Guinea, Sierra Leone, and Liberia from December 23, 2013, to August 11, 2014. Data are from the WHO. Briand S et al. N Engl J Med DOI: /NEJMp

54
Frieden TR et al. N Engl J Med 2014. DOI: 10.1056/NEJMp1409903
Elements of the Global Health Security Agenda and Their Application to the Ebola Outbreak. Frieden TR et al. N Engl J Med DOI: /NEJMp

55
Wolz A. N Engl J Med 2014. DOI: 10.1056/NEJMp1410179
MSF Staff Members Lead a Young Patient with Suspected Ebola into the Case-Management Center. MSF Staff Members Lead a Young Patient with Suspected Ebola into the Case-Management Center. Photo by Sylvain Cherkaoui/Cosmos/Médecins sans Frontières/Redux. Wolz A. N Engl J Med DOI: /NEJMp

57
William Osler (Band Head, 1849 - Oxford, 1919)
")
58
Chauvinismo en Medicina1902.
“Arrancar a la naturaleza los secretos que han dejado perplejos a los filósofos de todos los tiempos, seguir el rastro hasta su origen a las causas de la enfermedad, correlacionar los inmensos almacenes de conocimientos para que puedan estar fácilmente disponibles para la prevención y cura de la enfermedad. Esas son nuestras ambiciones” William Osler. Chauvinismo en Medicina1902.

59
El Hospital como Facultad. Osler 1903
Recomienda la asignación de los estudiantes de tercer curso a las salas de consultas externas y quirúrgicas para la exploración rutinaria de pacientes bajo supervisión experta. El cuarto curso debe dedicarse a trabajar en las salas, no a recibir clases en las salas. Deben encargarse de 4 o 5 camas y ayudar periódicamente en la visita por todas las salas.

60
Formación médica siglo XXI
Formación de grado - 6 años Formación de postgrado Formación continuada

61
Formación médica siglo XXI
Formación de grado - 6 años Formación de postgrado años. Troncalidad RD 639/2014 de 25/07 Formación continuada

64
Patología General. Introducción a la Medicina Clínica
Parte I: Generalidades Parte II: Respiratorio Parte III: Nefrología Parte IV: Metabolismo

65
Patología General. Introducción a la Medicina Clínica
Parte V: Endocrino Parte VI: Hematología Parte VII: Cardiocirculatorio Parte VIII: Digestivo Parte IX: Neurología

66
CLÍNICA actividad que realiza el médico junto al enfermo
- DIAGNÓSTICO Recogida de Datos Análisis e interpretación de los datos Identificación de Síndrome Diagnóstico diferencial Identificación etiología - PRONÓSTICO - TRATAMIENTO

67
Clases y Seminarios Contenidos Clases Lunes Generalidades Metabolismo Endocrino Digestivo Clases Martes Nefrología Hematología Neurología Clases Jueves Neumología Cardiología Seminarios 10 grupos 1 semana . Lunes-Viernes. Semiología Exploración física Exploraciones complementarias

68
Evaluación Número de Preguntas Respuesta múltiple Errores ( -0.25) ELIMINA PARCIAL ELIMINATORIO PARTES I,II,III,IV 40 70% aciertos (28) Mínimo 50% cada parcial (5) Examen final PARTES NO ELIMINADAS EN PARCIAL + PARTES V,VI,VII,VIII,IX Semiología 100 50% aciertos (50) Mínimo 35% cada parcial (3.5)
 Mínimo 50% cada parcial (5) Examen final. PARTES NO ELIMINADAS. EN PARCIAL. + PARTES V,VI,VII,VIII,IX. Semiología % aciertos (50) Mínimo 35% cada parcial (3.5)")
69
Genética en Patología General
Remigio Cordero Torres Badajoz 14 de Septiembre de 2015

70
FENOTIPO = GENOTIPO + AMBIENTE
Resultado de la constitución genetica heredada más los factores ambientales Enfermedad: Resultado de la interacción entre la constitución genética y el entorno

71
MUTACIONES Cambios en la secuencia de DNA de un gen
Mutación: referencia a genotipo Mutante: referencia a fenotipo Mutación GERMINAL: Afecta a linea germinal. Transmisible a sucesivas generaciones Mutación SOMÁTICA: Afecta linea no germinal. Capacidad para originar clones (cels troncales). No hereditarias. Pueden originar tumores.
. No hereditarias. Pueden originar tumores.")
72
Efectos de una mutación
Sobre la producción de una proteína Disminuir o detener su producción Aumentar su producción Alterar su función (su secreción, localización o interacción con otra proteína)
")
73
Functional Analysis of Hb JP
Figure 2. Functional Analysis of Hb JP. Dialyzed hemolysate prepared from the patient's blood had a higher partial pressure of oxygen (PO2) at 50 percent dissociation (P50) than wild-type HbA (Panel A). Whereas the P50 of purified Hb JP was only slightly higher than that of HbS alone, the difference increased in the presence of 3.0 mM 2,3-diphosphoglycerate (DPG) (Panel B). Tetramer formation for Hb JP was within the normal range and not significantly different from that of HbS (Panel C). Insets show the data replotted to obtain the dissociation constant (kD). T denotes the extent of tetramer formation, expressed as a percentage. Panel D shows a proposed mechanism for the reduced oxygen affinity of Hb JP. The phenylalanine substitution at position 68 of the {beta} chain was modeled according to the coordinates from normal HbA to assess how it would affect the binding of oxygen (red ball). The oxy-HbA backbone was not changed in this model, but the leucine residue at position 68 was replaced with phenylalanine. Side chains surrounding the heme moiety (histidine at position 63, lysine at 66, and valine at 67) are shown for reference (purple). Additional side chains shown for reference include tryptophan at position 15 (yellow) and leucine at positions 110 and 114 (orange). The solid red lines between the bulky mutant phenylalanine residue at position 68 (green) and nearby chains (valine at positions 20 and 23 and glycine at position 24 [blue]) represent highly unfavorable contacts. A rearrangement would probably occur in order to compensate for these adverse contacts. When the same analysis of phenylalanine substitution is performed with deoxy-HbA coordinates (not shown), the unfavorable interactions are much less severe. Hence, the deoxy structure would be preferred, suggesting that oxygen binding would be weaker (i.e., there would be a rightward shift of the hemoglobin dissociation curve). Amino acid residues are labeled with their single-letter codes. Colors denote the alpha helix (A, B, E, or G) of {beta} globin in which the amino acids are located. Geva A et al. N Engl J Med 2004;351:
 at 50 percent dissociation (P50) than wild-type HbA (Panel A). Whereas the P50 of purified Hb JP was only slightly higher than that of HbS alone, the difference increased in the presence of 3.0 mM 2,3-diphosphoglycerate (DPG) (Panel B). Tetramer formation for Hb JP was within the normal range and not significantly different from that of HbS (Panel C). Insets show the data replotted to obtain the dissociation constant (kD). T denotes the extent of tetramer formation, expressed as a percentage. Panel D shows a proposed mechanism for the reduced oxygen affinity of Hb JP. The phenylalanine substitution at position 68 of the {beta} chain was modeled according to the coordinates from normal HbA to assess how it would affect the binding of oxygen (red ball). The oxy-HbA backbone was not changed in this model, but the leucine residue at position 68 was replaced with phenylalanine. Side chains surrounding the heme moiety (histidine at position 63, lysine at 66, and valine at 67) are shown for reference (purple). Additional side chains shown for reference include tryptophan at position 15 (yellow) and leucine at positions 110 and 114 (orange). The solid red lines between the bulky mutant phenylalanine residue at position 68 (green) and nearby chains (valine at positions 20 and 23 and glycine at position 24 [blue]) represent highly unfavorable contacts. A rearrangement would probably occur in order to compensate for these adverse contacts. When the same analysis of phenylalanine substitution is performed with deoxy-HbA coordinates (not shown), the unfavorable interactions are much less severe. Hence, the deoxy structure would be preferred, suggesting that oxygen binding would be weaker (i.e., there would be a rightward shift of the hemoglobin dissociation curve). Amino acid residues are labeled with their single-letter codes. Colors denote the alpha helix (A, B, E, or G) of {beta} globin in which the amino acids are located. Geva A et al. N Engl J Med 2004;351:")
74
Desaturation of Normal and Variant Hemoglobins during Passage from Artery to Vein
Figure. Desaturation of Normal and Variant Hemoglobins during Passage from Artery to Vein. Hemoglobins lose oxygen during the journey from artery to vein. Cells with HbS sickle only when desaturated. The likelihood that they will obstruct the vessel depends on the concentration of HbS and the extent of desaturation. Benz E. N Engl J Med 2004;351:

75
Hypothetical Mechanisms of Clinical Subphenotypes of Sickle Cell Disease
Figure 1. Hypothetical Mechanisms of Clinical Subphenotypes of Sickle Cell Disease. It is hypothesized that many of the complications of sickle cell disease can be divided into two overlapping subtypes, each driven by distinct mechanisms. Cutaneous leg ulceration, priapism, pulmonary hypertension, sudden death, and stroke are associated with low steady-state hemoglobin (Hb) levels and an increased rate of intravascular hemolysis, shown on the left side of the figure. These vasculopathic complications probably result from endothelial dysfunction, mediated by both inactivation of nitric oxide (NO) by free-plasma hemoglobin and vascular reactive oxygen species as well as arginine (Arg) catabolism by plasma arginase. This process of hemolysis-associated endothelial dysfunction may also cause hemostatic activation and intimal and smooth-muscle proliferation. Such clinical complications as vaso-occlusive pain crisis, the acute chest syndrome, avascular necrosis of bones, and retinal vasculopathy are associated with high steady-state leukocyte counts and high hemoglobin levels. These complications are likely to result from obstruction of capillaries and postcapillary venules by erythrocytes containing polymerized hemoglobin S and by leukocytes (a monocyte is shown), as shown on the right side of the figure. ET-1 denotes endothelin 1, NOS nitric oxide synthase, O2- superoxide, VCAM-1 vascular-cell adhesion molecule 1, and XO xanthine oxidase. Gladwin M and Vichinsky E. N Engl J Med 2008;359:
 levels and an increased rate of intravascular hemolysis, shown on the left side of the figure. These vasculopathic complications probably result from endothelial dysfunction, mediated by both inactivation of nitric oxide (NO) by free-plasma hemoglobin and vascular reactive oxygen species as well as arginine (Arg) catabolism by plasma arginase. This process of hemolysis-associated endothelial dysfunction may also cause hemostatic activation and intimal and smooth-muscle proliferation. Such clinical complications as vaso-occlusive pain crisis, the acute chest syndrome, avascular necrosis of bones, and retinal vasculopathy are associated with high steady-state leukocyte counts and high hemoglobin levels. These complications are likely to result from obstruction of capillaries and postcapillary venules by erythrocytes containing polymerized hemoglobin S and by leukocytes (a monocyte is shown), as shown on the right side of the figure. ET-1 denotes endothelin 1, NOS nitric oxide synthase, O2- superoxide, VCAM-1 vascular-cell adhesion molecule 1, and XO xanthine oxidase. Gladwin M and Vichinsky E. N Engl J Med 2008;359:")
76
The Thyrotropin Receptor
Figure 1. The Thyrotropin Receptor. The location of constitutively activating mutations1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16 and inactivating mutations15,17,18 of the thyrotropin-receptor gene is shown, as is the location of somatic mutations found in thyroid carcinomas.10,19,20 At some locations, several different amino acid substitutions have been described. All gain-of-function mutations are in exon 10 except Ser281Asn/Thr, which is in exon 9. Gain-of-function mutations are denoted by circles in the case of hyperfunctioning thyroid adenomas, squares in the case of familial autosomal dominant hyperthyroidism, diamonds in the case of sporadic congenital hyperthyroidism, and octagons in the case of thyroid carcinomas. Loss-of-function mutations are denoted by triangles. Letters indicate the amino acid in the wild-type receptor. The asterisk and double asterisk indicate deletions resulting in a gain of function in hyperfunctioning thyroid adenomas. Paschke, R. et al. N Engl J Med 1997;337:

77
Components of Myocyte Cytoarchitecture (Panel A) and Mutations Causing Dilated Cardiomyopathy and Conduction-System Disease or Autosomal Dominant Emery-Dreifuss Muscular Dystrophy (Panel B) Figure 1. Components of Myocyte Cytoarchitecture (Panel A) and Mutations Causing Dilated Cardiomyopathy and Conduction-System Disease or Autosomal Dominant Emery-Dreifuss Muscular Dystrophy (Panel B). Mutations in the rod domain of the lamin A/C gene cause isolated dilated cardiomyopathy and conduction-system disease, presumably through perturbed interactions with nuclear or cytoplasmic constituents (Panel A). Other cytoskeletal molecules implicated in the pathophysiology of human dilated cardiomyopathy include actin, dystrophin, and the dystrophin-associated glycoprotein complex.12,23,24,25,26,27,28 Interactions between lamins A and C and cytoskeletal or sarcomere proteins are unknown. Conduction-system disease is a common feature of Emery-Dreifuss muscular dystrophy caused by defects in the head or tail domain of the lamin gene or by emerin mutations. Mutations causing dilated cardiomyopathy and conduction-system disease or autosomal dominant Emery-Dreifuss muscular dystrophy are distributed in distinct domains of the lamin dimer (Panel B). Lamins A and C have identical structures throughout the amino-terminal head (NH3), {alpha}-helical rod domain, and proximal carboxyl-terminal tail (COOH), but they differ in their distal amino acids (lamin A is shown in gray, and lamin C is shown in black). Mutations in the rod domain (Arg60Gly, Leu85Arg, Asn195Lys, and Glu203Gly) cause dilated cardiomyopathy and conduction-system disease without skeletal myopathy; the mutation at the carboxyl terminal (Arg571Ser) is associated with subclinical skeletal-muscle disease. Mutations that cause Emery-Dreifuss muscular dystrophy (Gln6Stop, Arg453Trp, Arg527Pro, and Leu530Pro) do not affect the {alpha}-helical rod domain. Fatkin, D. et al. N Engl Med 1999;341: J
 and Mutations Causing Dilated Cardiomyopathy and Conduction-System Disease or Autosomal Dominant Emery-Dreifuss Muscular Dystrophy (Panel B). Mutations in the rod domain of the lamin A/C gene cause isolated dilated cardiomyopathy and conduction-system disease, presumably through perturbed interactions with nuclear or cytoplasmic constituents (Panel A). Other cytoskeletal molecules implicated in the pathophysiology of human dilated cardiomyopathy include actin, dystrophin, and the dystrophin-associated glycoprotein complex.12,23,24,25,26,27,28 Interactions between lamins A and C and cytoskeletal or sarcomere proteins are unknown. Conduction-system disease is a common feature of Emery-Dreifuss muscular dystrophy caused by defects in the head or tail domain of the lamin gene or by emerin mutations. Mutations causing dilated cardiomyopathy and conduction-system disease or autosomal dominant Emery-Dreifuss muscular dystrophy are distributed in distinct domains of the lamin dimer (Panel B). Lamins A and C have identical structures throughout the amino-terminal head (NH3), {alpha}-helical rod domain, and proximal carboxyl-terminal tail (COOH), but they differ in their distal amino acids (lamin A is shown in gray, and lamin C is shown in black). Mutations in the rod domain (Arg60Gly, Leu85Arg, Asn195Lys, and Glu203Gly) cause dilated cardiomyopathy and conduction-system disease without skeletal myopathy; the mutation at the carboxyl terminal (Arg571Ser) is associated with subclinical skeletal-muscle disease. Mutations that cause Emery-Dreifuss muscular dystrophy (Gln6Stop, Arg453Trp, Arg527Pro, and Leu530Pro) do not affect the {alpha}-helical rod domain. Fatkin, D. et al. N Engl Med 1999;341: J.")
78
PATOGENIA ERRORES CONGÉNITOS DEL METABOLISMO
MUTACIÓN FALLO ENZIMÁTICO MECANISMOS ENFERMEDAD: AUSENCIA PRODUCTO FINAL ACUMULACIÓN PRODUCTO PREVIO DERIVACIÓN ANORMAL CAMINO METABÓLICO ALTERNATIVO RUPTURA MECANISMO REGULADOR AL ALTERAR CANTIDAD METABOLISMO

79
PORFIRIAS:Bloqueo Precoz
PBG DESAMINASA PORFIRIA AGUDA INTERMITENTE ALA PBG HEM

80
Efectos de una mutación
Sobre la producción de una proteína Disminuir o detener su producción Aumentar su producción Alterar su función (su secreción, localización o interacción con otra proteína) NO TODAS LAS MUTACIONES SON PERJUDICIALES (CCR5)
 NO TODAS LAS MUTACIONES SON PERJUDICIALES (CCR5)")
81
The Life Cycle of Human Immunodeficiency Virus Type 1 (HIV-1), Showing Potential Targets for Antiretroviral Therapy Figure 1. The Life Cycle of Human Immunodeficiency Virus Type 1 (HIV-1), Showing Potential Targets for Antiretroviral Therapy. HIV-1 binds to receptors on the cell surface, undergoes membrane fusion, and then releases copies of the RNA genome into the cytoplasm. After successful invasion of the cell, the viral reverse-transcriptase enzyme transcribes single-stranded viral RNA into double-stranded DNA that can be integrated into the genetic material of the human host. Reverse-transcriptase inhibitors were the first agents approved for the treatment of HIV-1; currently available inhibitors of this enzyme are nucleoside antagonists (zidovudine, didanosine, zalcitabine, lamivudine, stavudine, abacavir, and combined formulations), nonnucleoside competitive inhibitors (nevirapine, delavirdine, and efavirenz), and one nucleotide analogue (tenofovir). The viral integrase enzyme is required for the integration of proviral DNA into the host genome before replication. Investigational integrase inhibitors are currently in early clinical trials. When the infected cell synthesizes new protein, integrated proviral DNA is also translated into the protein building blocks of new viral progeny. The viral components then assemble on the cell surface and bud out as immature viral particles. The final maturation of newly formed viruses requires the HIV-1 protease to digest larger components into the intricate pieces that make up an infectious virion. Several protease inhibitors (ritonavir, indinavir, nelfinavir, amprenavir, lopinavir-ritonavir, and two formulations of saquinavir) are currently in clinical use. Kilby J and Eron J. N Engl J Med 2003;348:
, Showing Potential Targets for Antiretroviral Therapy. HIV-1 binds to receptors on the cell surface, undergoes membrane fusion, and then releases copies of the RNA genome into the cytoplasm. After successful invasion of the cell, the viral reverse-transcriptase enzyme transcribes single-stranded viral RNA into double-stranded DNA that can be integrated into the genetic material of the human host. Reverse-transcriptase inhibitors were the first agents approved for the treatment of HIV-1; currently available inhibitors of this enzyme are nucleoside antagonists (zidovudine, didanosine, zalcitabine, lamivudine, stavudine, abacavir, and combined formulations), nonnucleoside competitive inhibitors (nevirapine, delavirdine, and efavirenz), and one nucleotide analogue (tenofovir). The viral integrase enzyme is required for the integration of proviral DNA into the host genome before replication. Investigational integrase inhibitors are currently in early clinical trials. When the infected cell synthesizes new protein, integrated proviral DNA is also translated into the protein building blocks of new viral progeny. The viral components then assemble on the cell surface and bud out as immature viral particles. The final maturation of newly formed viruses requires the HIV-1 protease to digest larger components into the intricate pieces that make up an infectious virion. Several protease inhibitors (ritonavir, indinavir, nelfinavir, amprenavir, lopinavir-ritonavir, and two formulations of saquinavir) are currently in clinical use. Kilby J and Eron J. N Engl J Med 2003;348:")
82
HIV-1-Binding Events and Potential Sites of Action for Various Viral-Entry Inhibitors
Figure 2. HIV-1-Binding Events and Potential Sites of Action for Various Viral-Entry Inhibitors. HIV-1 is covered by a lipid bilayer derived from host-cell membranes. Incorporated into this bilayer are viral glycoproteins as well as host adhesion molecules that may play a part in attachment to target cells. The viral-entry process consists of a series of coordinated interactions -- binding to two different receptors (Panel A) and membrane fusion (Panel B). The viral envelope glycoproteins are synthesized as a single polyprotein that assembles into a trimer and then is broken down by host protease into surface glycoprotein subunits (gp120) and transmembrane glycoprotein subunits (gp41). Each gp120 monomer is a complex, folded structure, consisting of a series of variable loops formed by disulfide bonds, with noncontiguous segments brought together to form three-dimensional binding sites for the CD4 receptor and a chemokine receptor (either CCR5 or CXCR4). Initial binding of gp120 to CD4 (Panel A) might be blocked by soluble CD4 decoys, monoclonal antibodies against sequences on gp120 or CD4, or other small-molecular inhibitors. After CD4 binding, each gp120 undergoes a conformational change exposing the region that will bind to a seven-transmembrane chemokine receptor. Viral isolates have varying affinities for CCR5 or CXCR4 receptors. Binding of the chemokine coreceptors might be inhibited by natural ligands of these receptors or their derivatives, small-molecule inhibitors, monoclonal antibodies directed at the interacting sites, or down-regulation of receptor expression. It is hypothesized that binding of both the CD4 and chemokine receptors shifts away the steric hindrance of the heavily glycosylated gp120, allowing the gp41 segment to mediate membrane fusion and entry (Panel B). Kilby J and Eron J. N Engl J Med 2003;348:
 and membrane fusion (Panel B). The viral envelope glycoproteins are synthesized as a single polyprotein that assembles into a trimer and then is broken down by host protease into surface glycoprotein subunits (gp120) and transmembrane glycoprotein subunits (gp41). Each gp120 monomer is a complex, folded structure, consisting of a series of variable loops formed by disulfide bonds, with noncontiguous segments brought together to form three-dimensional binding sites for the CD4 receptor and a chemokine receptor (either CCR5 or CXCR4). Initial binding of gp120 to CD4 (Panel A) might be blocked by soluble CD4 decoys, monoclonal antibodies against sequences on gp120 or CD4, or other small-molecular inhibitors. After CD4 binding, each gp120 undergoes a conformational change exposing the region that will bind to a seven-transmembrane chemokine receptor. Viral isolates have varying affinities for CCR5 or CXCR4 receptors. Binding of the chemokine coreceptors might be inhibited by natural ligands of these receptors or their derivatives, small-molecule inhibitors, monoclonal antibodies directed at the interacting sites, or down-regulation of receptor expression. It is hypothesized that binding of both the CD4 and chemokine receptors shifts away the steric hindrance of the heavily glycosylated gp120, allowing the gp41 segment to mediate membrane fusion and entry (Panel B). Kilby J and Eron J. N Engl J Med 2003;348:")
83
HIV-1-Binding Events and Potential Sites of Action for Various Viral-Entry Inhibitors
Figure 2. HIV-1-Binding Events and Potential Sites of Action for Various Viral-Entry Inhibitors. HIV-1 is covered by a lipid bilayer derived from host-cell membranes. Incorporated into this bilayer are viral glycoproteins as well as host adhesion molecules that may play a part in attachment to target cells. The viral-entry process consists of a series of coordinated interactions -- binding to two different receptors (Panel A) and membrane fusion (Panel B). The viral envelope glycoproteins are synthesized as a single polyprotein that assembles into a trimer and then is broken down by host protease into surface glycoprotein subunits (gp120) and transmembrane glycoprotein subunits (gp41). Each gp120 monomer is a complex, folded structure, consisting of a series of variable loops formed by disulfide bonds, with noncontiguous segments brought together to form three-dimensional binding sites for the CD4 receptor and a chemokine receptor (either CCR5 or CXCR4). Initial binding of gp120 to CD4 (Panel A) might be blocked by soluble CD4 decoys, monoclonal antibodies against sequences on gp120 or CD4, or other small-molecular inhibitors. After CD4 binding, each gp120 undergoes a conformational change exposing the region that will bind to a seven-transmembrane chemokine receptor. Viral isolates have varying affinities for CCR5 or CXCR4 receptors. Binding of the chemokine coreceptors might be inhibited by natural ligands of these receptors or their derivatives, small-molecule inhibitors, monoclonal antibodies directed at the interacting sites, or down-regulation of receptor expression. It is hypothesized that binding of both the CD4 and chemokine receptors shifts away the steric hindrance of the heavily glycosylated gp120, allowing the gp41 segment to mediate membrane fusion and entry (Panel B). Kilby J and Eron J. N Engl J Med 2003;348:
 and membrane fusion (Panel B). The viral envelope glycoproteins are synthesized as a single polyprotein that assembles into a trimer and then is broken down by host protease into surface glycoprotein subunits (gp120) and transmembrane glycoprotein subunits (gp41). Each gp120 monomer is a complex, folded structure, consisting of a series of variable loops formed by disulfide bonds, with noncontiguous segments brought together to form three-dimensional binding sites for the CD4 receptor and a chemokine receptor (either CCR5 or CXCR4). Initial binding of gp120 to CD4 (Panel A) might be blocked by soluble CD4 decoys, monoclonal antibodies against sequences on gp120 or CD4, or other small-molecular inhibitors. After CD4 binding, each gp120 undergoes a conformational change exposing the region that will bind to a seven-transmembrane chemokine receptor. Viral isolates have varying affinities for CCR5 or CXCR4 receptors. Binding of the chemokine coreceptors might be inhibited by natural ligands of these receptors or their derivatives, small-molecule inhibitors, monoclonal antibodies directed at the interacting sites, or down-regulation of receptor expression. It is hypothesized that binding of both the CD4 and chemokine receptors shifts away the steric hindrance of the heavily glycosylated gp120, allowing the gp41 segment to mediate membrane fusion and entry (Panel B). Kilby J and Eron J. N Engl J Med 2003;348:")
84
Efectos de una mutación
Sobre la producción de una proteína Disminuir o detener su producción Aumentar su producción Alterar su función (su secreción, localización o interacción con otra proteína) NO TODAS LAS MUTACIONES SON PERJUDICIALES (CCR5) Sobre las poblaciones Esencial para la vida. Produce individuos con variantes fenotípicas que pueden sobrevivir mejor a cambios ambientales
 NO TODAS LAS MUTACIONES SON PERJUDICIALES (CCR5) Sobre las poblaciones. Esencial para la vida. Produce individuos con variantes fenotípicas que pueden sobrevivir mejor a cambios ambientales.")
85
Antimalarial Drug Activity in the Life Cycle of Plasmodia
Figure 2. Antimalarial Drug Activity in the Life Cycle of Plasmodia. Tissue-stage schizonticides kill the asexual stages developing in the liver, including liver schizonts (all species) and quiescent hypnozoites (Plasmodium vivax and P. ovale), thus preventing primary or secondary attacks (relapses) of clinical malaria. Blood-stage schizonticides interrupt asexual schizogony (mitotic division) in red cells, preventing or terminating clinical attacks of malaria. Gametocytocides kill or sterilize sexual stages in the blood, thus preventing infection of mosquitoes and transmission of the disease. Another class of drugs, the sporontocides (which kill forms developing in the mosquito, including the sporozoites that infect humans), is not represented here, because none are available for clinical use. Baird J. N Engl J Med 2005;352:
 and quiescent hypnozoites (Plasmodium vivax and P. ovale), thus preventing primary or secondary attacks (relapses) of clinical malaria. Blood-stage schizonticides interrupt asexual schizogony (mitotic division) in red cells, preventing or terminating clinical attacks of malaria. Gametocytocides kill or sterilize sexual stages in the blood, thus preventing infection of mosquitoes and transmission of the disease. Another class of drugs, the sporontocides (which kill forms developing in the mosquito, including the sporozoites that infect humans), is not represented here, because none are available for clinical use. Baird J. N Engl J Med 2005;352:")
86
Functional Analysis of Hb JP
Figure 2. Functional Analysis of Hb JP. Dialyzed hemolysate prepared from the patient's blood had a higher partial pressure of oxygen (PO2) at 50 percent dissociation (P50) than wild-type HbA (Panel A). Whereas the P50 of purified Hb JP was only slightly higher than that of HbS alone, the difference increased in the presence of 3.0 mM 2,3-diphosphoglycerate (DPG) (Panel B). Tetramer formation for Hb JP was within the normal range and not significantly different from that of HbS (Panel C). Insets show the data replotted to obtain the dissociation constant (kD). T denotes the extent of tetramer formation, expressed as a percentage. Panel D shows a proposed mechanism for the reduced oxygen affinity of Hb JP. The phenylalanine substitution at position 68 of the {beta} chain was modeled according to the coordinates from normal HbA to assess how it would affect the binding of oxygen (red ball). The oxy-HbA backbone was not changed in this model, but the leucine residue at position 68 was replaced with phenylalanine. Side chains surrounding the heme moiety (histidine at position 63, lysine at 66, and valine at 67) are shown for reference (purple). Additional side chains shown for reference include tryptophan at position 15 (yellow) and leucine at positions 110 and 114 (orange). The solid red lines between the bulky mutant phenylalanine residue at position 68 (green) and nearby chains (valine at positions 20 and 23 and glycine at position 24 [blue]) represent highly unfavorable contacts. A rearrangement would probably occur in order to compensate for these adverse contacts. When the same analysis of phenylalanine substitution is performed with deoxy-HbA coordinates (not shown), the unfavorable interactions are much less severe. Hence, the deoxy structure would be preferred, suggesting that oxygen binding would be weaker (i.e., there would be a rightward shift of the hemoglobin dissociation curve). Amino acid residues are labeled with their single-letter codes. Colors denote the alpha helix (A, B, E, or G) of {beta} globin in which the amino acids are located. Geva A et al. N Engl J Med 2004;351:
 at 50 percent dissociation (P50) than wild-type HbA (Panel A). Whereas the P50 of purified Hb JP was only slightly higher than that of HbS alone, the difference increased in the presence of 3.0 mM 2,3-diphosphoglycerate (DPG) (Panel B). Tetramer formation for Hb JP was within the normal range and not significantly different from that of HbS (Panel C). Insets show the data replotted to obtain the dissociation constant (kD). T denotes the extent of tetramer formation, expressed as a percentage. Panel D shows a proposed mechanism for the reduced oxygen affinity of Hb JP. The phenylalanine substitution at position 68 of the {beta} chain was modeled according to the coordinates from normal HbA to assess how it would affect the binding of oxygen (red ball). The oxy-HbA backbone was not changed in this model, but the leucine residue at position 68 was replaced with phenylalanine. Side chains surrounding the heme moiety (histidine at position 63, lysine at 66, and valine at 67) are shown for reference (purple). Additional side chains shown for reference include tryptophan at position 15 (yellow) and leucine at positions 110 and 114 (orange). The solid red lines between the bulky mutant phenylalanine residue at position 68 (green) and nearby chains (valine at positions 20 and 23 and glycine at position 24 [blue]) represent highly unfavorable contacts. A rearrangement would probably occur in order to compensate for these adverse contacts. When the same analysis of phenylalanine substitution is performed with deoxy-HbA coordinates (not shown), the unfavorable interactions are much less severe. Hence, the deoxy structure would be preferred, suggesting that oxygen binding would be weaker (i.e., there would be a rightward shift of the hemoglobin dissociation curve). Amino acid residues are labeled with their single-letter codes. Colors denote the alpha helix (A, B, E, or G) of {beta} globin in which the amino acids are located. Geva A et al. N Engl J Med 2004;351:")
87
Desaturation of Normal and Variant Hemoglobins during Passage from Artery to Vein
Figure. Desaturation of Normal and Variant Hemoglobins during Passage from Artery to Vein. Hemoglobins lose oxygen during the journey from artery to vein. Cells with HbS sickle only when desaturated. The likelihood that they will obstruct the vessel depends on the concentration of HbS and the extent of desaturation. Benz E. N Engl J Med 2004;351:

88
Platelet Activation in Falciparum Malaria: Two Models
Figure 1. Platelet Activation in Falciparum Malaria: Two Models. McMorran et al.3 recently reported that erythrocytes infected with Plasmodium falciparum activate platelets and induce the formation of platelet-erythrocyte complexes that result in the killing of parasites (Panel A). Inhibition of platelet activation through the use of aspirin (or platelet depletion by means of genetic manipulation) limited, but did not abolish, erythrocyte infestation with a similar, but distinct, parasite -- P. chabaudi -- in mice. Thus, if these observations can be extrapolated to P. falciparum infection in humans, aspirin treatment for pyrexia in the early stages of malaria may be undesirable. In contrast, Srivastava et al.4 described a setting in which platelet inhibition with aspirin might be desirable (Panel B). They showed -- using mice infected with another related parasite, P. berghei -- that platelet factor 4 (Pf4) released by platelets (after activation by dysfunctional endothelium) facilitates an interaction between endothelial cells and erythrocytes, exacerbation of cerebral malaria, and, potentially, vascular occlusion. Greenbaum D and FitzGerald G. N Engl J Med 2009;361:
. Inhibition of platelet activation through the use of aspirin (or platelet depletion by means of genetic manipulation) limited, but did not abolish, erythrocyte infestation with a similar, but distinct, parasite -- P. chabaudi -- in mice. Thus, if these observations can be extrapolated to P. falciparum infection in humans, aspirin treatment for pyrexia in the early stages of malaria may be undesirable. In contrast, Srivastava et al.4 described a setting in which platelet inhibition with aspirin might be desirable (Panel B). They showed -- using mice infected with another related parasite, P. berghei -- that platelet factor 4 (Pf4) released by platelets (after activation by dysfunctional endothelium) facilitates an interaction between endothelial cells and erythrocytes, exacerbation of cerebral malaria, and, potentially, vascular occlusion. Greenbaum D and FitzGerald G. N Engl J Med 2009;361:")
89
A 23-year-old man with sickle cell disease was admitted after reporting fever and chills
A 23-year-old man with sickle cell disease was admitted after reporting fever and chills. He had emigrated from West Africa three months earlier. His temperature was 38.2{degrees}C, and his other vital signs were normal. His spleen was not palpable. The hemoglobin level was 8.7 g per deciliter. Electrophoresis showed a single band of hemoglobin S consistent with the presence of hemoglobin SS sickle cell disease. Examination of a blood smear revealed a few sickle cells (arrowhead) and rare red cells with Plasmodium falciparum (arrow); the patient had had no history of malaria. Although hemoglobin S is considered to be protective against P. falciparum, this is not always the case. Boctor F and Uehlinger J. N Engl J Med 2002;347:e1
 and rare red cells with Plasmodium falciparum (arrow); the patient had had no history of malaria. Although hemoglobin S is considered to be protective against P. falciparum, this is not always the case. Boctor F and Uehlinger J. N Engl J Med 2002;347:e1.")
90
Risk of Plasmodium falciparum Malaria Worldwide
Figure 1. Risk of Plasmodium falciparum Malaria Worldwide. Adapted from the Public Health Mapping Group, Communicable Diseases, World Health Organization, November 2002. Baird J. N Engl J Med 2005;352:

91
Tipos de mutaciones CLASIFICACIÓN MORFOLÓGICA
PUNTUALES Transiciones: Purina-Purina (A-G, G-A) Pirimidina- Pirimidina (C-T,T-C) Transversiones: Purina-Pirimidina ALTERACIÓN NÚMERO DE BASES Delección. Inserción. Expansión de repetición de tripletes.
 Pirimidina- Pirimidina (C-T,T-C) Transversiones: Purina-Pirimidina. ALTERACIÓN NÚMERO DE BASES. Delección. Inserción. Expansión de repetición de tripletes.")
92
Examples of Point Mutations
Figure 3. Examples of Point Mutations. Panel A shows the normal sequence of DNA from one exon and the protein product it encodes. Panel B shows a silent mutation, Panel C a conservative missense mutation (serine and threonine have very similar structures), Panel D a nonconservative missense mutation (serine and proline have very different structures), Panel E a nonsense mutation, and Panel F a frame-shift mutation. In Panel F, the insertion of a single G throws off the reading frame, so that all amino acids downstream are changed radically. Guttmacher, A. E. et al. N Engl J Med 2002;347:
, Panel D a nonconservative missense mutation (serine and proline have very different structures), Panel E a nonsense mutation, and Panel F a frame-shift mutation. In Panel F, the insertion of a single G throws off the reading frame, so that all amino acids downstream are changed radically. Guttmacher, A. E. et al. N Engl J Med 2002;347:")
93
Examples of Point Mutations
Figure 3. Examples of Point Mutations. Panel A shows the normal sequence of DNA from one exon and the protein product it encodes. Panel B shows a silent mutation, Panel C a conservative missense mutation (serine and threonine have very similar structures), Panel D a nonconservative missense mutation (serine and proline have very different structures), Panel E a nonsense mutation, and Panel F a frame-shift mutation. In Panel F, the insertion of a single G throws off the reading frame, so that all amino acids downstream are changed radically. Guttmacher, A. E. et al. N Engl J Med 2002;347:
, Panel D a nonconservative missense mutation (serine and proline have very different structures), Panel E a nonsense mutation, and Panel F a frame-shift mutation. In Panel F, the insertion of a single G throws off the reading frame, so that all amino acids downstream are changed radically. Guttmacher, A. E. et al. N Engl J Med 2002;347:")
94
Examples of Point Mutations
Figure 3. Examples of Point Mutations. Panel A shows the normal sequence of DNA from one exon and the protein product it encodes. Panel B shows a silent mutation, Panel C a conservative missense mutation (serine and threonine have very similar structures), Panel D a nonconservative missense mutation (serine and proline have very different structures), Panel E a nonsense mutation, and Panel F a frame-shift mutation. In Panel F, the insertion of a single G throws off the reading frame, so that all amino acids downstream are changed radically. Guttmacher, A. E. et al. N Engl J Med 2002;347:
, Panel D a nonconservative missense mutation (serine and proline have very different structures), Panel E a nonsense mutation, and Panel F a frame-shift mutation. In Panel F, the insertion of a single G throws off the reading frame, so that all amino acids downstream are changed radically. Guttmacher, A. E. et al. N Engl J Med 2002;347:")
95
Examples of Point Mutations
Figure 3. Examples of Point Mutations. Panel A shows the normal sequence of DNA from one exon and the protein product it encodes. Panel B shows a silent mutation, Panel C a conservative missense mutation (serine and threonine have very similar structures), Panel D a nonconservative missense mutation (serine and proline have very different structures), Panel E a nonsense mutation, and Panel F a frame-shift mutation. In Panel F, the insertion of a single G throws off the reading frame, so that all amino acids downstream are changed radically. Guttmacher, A. E. et al. N Engl J Med 2002;347:
, Panel D a nonconservative missense mutation (serine and proline have very different structures), Panel E a nonsense mutation, and Panel F a frame-shift mutation. In Panel F, the insertion of a single G throws off the reading frame, so that all amino acids downstream are changed radically. Guttmacher, A. E. et al. N Engl J Med 2002;347:")
96
Examples of Point Mutations
Figure 3. Examples of Point Mutations. Panel A shows the normal sequence of DNA from one exon and the protein product it encodes. Panel B shows a silent mutation, Panel C a conservative missense mutation (serine and threonine have very similar structures), Panel D a nonconservative missense mutation (serine and proline have very different structures), Panel E a nonsense mutation, and Panel F a frame-shift mutation. In Panel F, the insertion of a single G throws off the reading frame, so that all amino acids downstream are changed radically. Guttmacher, A. E. et al. N Engl J Med 2002;347:
, Panel D a nonconservative missense mutation (serine and proline have very different structures), Panel E a nonsense mutation, and Panel F a frame-shift mutation. In Panel F, the insertion of a single G throws off the reading frame, so that all amino acids downstream are changed radically. Guttmacher, A. E. et al. N Engl J Med 2002;347:")
97
Tipos de mutación Normal THE ONE BIG FLY HAD ONE RED EYE
Missense THI ONE BIG FLY HAD ONE RED EYE Nonsense THE ONE BIG Stop Frameshift THE ONE IBI GFL YHA DON ERE DEY Delección THE ONE BIG HAD ONE RED EYE Inserción THE ONE BIG WET FLY HAD ONE RED Duplicación THE ONE BIG FLY FLY HAD ONE RED

98
Tipos de Mutaciones CLASIFICACIÓN FUNCIONAL
SILENCIOSAS No codifican ni control. Mismo Aa (CCA,CCU: Prol) DE CAMBIO DE SENTIDO Limitado a un triplete; sustitución SIN SENTIDO Inicio AUG, Terminación UAG, UGA, UAA DE ELEMENTOS DE CONTROL Promotor- intensificador DE CAMBIO DE ENCUADRE (Frame-shift) TACTACTAC: valina ACTACTACT: leucina DE EXPANSIÓN DE TRIPLETES. Anticipación.
 DE CAMBIO DE SENTIDO. Limitado a un triplete; sustitución. SIN SENTIDO. Inicio AUG, Terminación UAG, UGA, UAA. DE ELEMENTOS DE CONTROL. Promotor- intensificador. DE CAMBIO DE ENCUADRE (Frame-shift) TACTACTAC: valina. ACTACTACT: leucina. DE EXPANSIÓN DE TRIPLETES. Anticipación.")
99
Tipos de mutación. Expansión de tripletes
Normal THE ONE BIG FLY HAD ONE RED EYE Duplicación THE ONE BIG FLY FLY HAD ONE RED Expansión de tripletes. ANTICIPACIÓN Generación 2 THE ONE BIG FLY FLY FLY HAD ONE RED EYE Generación 3 THE ONE BIG FLY FLY FLY FLY HAD ONE RED EYE

100
TIPOS DE MUTACIONES MUTACIÓN ESPONTANEA MUTACIÓN INDUCIDA
Condiciones ambientales normales MUTACIÓN INDUCIDA Condiciones ambientales específicas AGENTE MUTAGÉNICO Acelerador tasa espontanea mutación. Tasa: para un gen determinado y por generación (20 años)
")
101
TASAS MUTACIÓN ESPONTANEA
En unicelulares se establece en cultivo En humanos se deduce de la incidencia de nuevas mutaciones dominantes ACONDROPLASIA: 10/millón de gametos HUNTINGTON: 1/millón de gametos NEUROFIBROMATOSIS: 100/millón gametos OSTEOGENESIS IMPERFECTA: 10/millón gm RETINOBLASTOMA: 5-12/millón gametos DISTROFIA MUSC DUCHENNE: 100/mill gmt

102
FACTORES CONDICIONANTES MUTACIÓN ESPONTANEA
TAMAÑO GEN Mayor probabilidad en genes mayores TIPO SECUENCIA DE BASES Repetición de bases, 5-metilcitosina INTRONES Más probabilidad error mutación para procesamiento

103
AGENTES MUTAGÉNICOS (II)
QUÍMICOS MEDICAMENTOS (CFM, MIT C, MTX) PESTICIDAS CONSERVANTES Y ADITIVOS INDUSTRIALES. Benceno. Cloruro Vinilo. COSMÉTICOS TABACO AFLATOXINA MECANISMO: Alquilantes: combinación con ADN: ADUCTOS.
 PESTICIDAS. CONSERVANTES Y ADITIVOS. INDUSTRIALES. Benceno. Cloruro Vinilo. COSMÉTICOS. TABACO. AFLATOXINA. MECANISMO: Alquilantes: combinación con ADN: ADUCTOS.")
104
MUTACIONES INDUCIDAS AGENTES MUTAGÉNICOS (I)
RADIACIONES Aumento tasa con intensidad ionización Dosis duplicación tasa en humanos: 169 rads Exposiciones acumulativas Hiroshima 1945, Chernobyl 1986 Cels troncales tejidos hematopoyéticos Epitelios de revestimiento Otros tejidos de alto índice mitótico Mecanismo: delecciones, roturas

105
Gearhart J et al. N Engl J Med 2007;357:1469-1472.
Induction of Pluripotent Stem Cells through Retroviral Transduction. Gearhart J et al. N Engl J Med 2007;357: Induction of Pluripotent Stem Cells through Retroviral Transduction. Retrovirally encoded transcription factor genes were introduced into mouse embryonic and adult fibroblasts. After integration and expression of the transgenes, the fibroblasts were reprogrammed to pluripotency.

106
Mummery C. N Engl J Med 2011;364:2160-2162.
Genetic Effects of Reprogramming Cells for Pluripotency. Figure 1. Genetic Effects of Reprogramming Cells for Pluripotency. Genetic lesions arise during reprogramming of fibroblasts to pluripotency (thus generating human induced pluripotent stem [hiPS] cells) and during prolonged culture of both hiPS cells and human embryonic stem (hES) cells. Recent studies show that in addition to gross chromosomal changes that occur during prolonged culture of hiPS and hES cells (e.g., duplication of parts of chromosomes 12 and 20), gene copy-number variations and point mutations can be induced during the reprogramming of somatic cells into hiPS cells, resulting in many more DNA lesions (by up to a factor of 10) in hiPS cells than in the somatic cells from which they are derived.1– 5 During prolonged culture, the frequency with which these new mutations are detected decreases. Mummery C. N Engl J Med 2011;364:
 and during prolonged culture of both hiPS cells and human embryonic stem (hES) cells. Recent studies show that in addition to gross chromosomal changes that occur during prolonged culture of hiPS and hES cells (e.g., duplication of parts of chromosomes 12 and 20), gene copy-number variations and point mutations can be induced during the reprogramming of somatic cells into hiPS cells, resulting in many more DNA lesions (by up to a factor of 10) in hiPS cells than in the somatic cells from which they are derived.1– 5 During prolonged culture, the frequency with which these new mutations are detected decreases. Mummery C. N Engl J Med 2011;364:")
107
Haruko Obokata – Yoshiki Sasai
Haruko Obokata – Yoshiki Sasai . Células Stap (stimulus-triggered acquisition pluripotency ) Artículo publicado en Nature en Enero 2014 Obokata dimite en Diciembre de 2014
 Artículo publicado en Nature en Enero Obokata dimite en Diciembre de")
108
MUTACIONES Cambios en la secuencia de DNA de un gen
Mutación: referencia a genotipo Mutante: referencia a fenotipo Mutación GERMINAL: Afecta a linea germinal. Transmisible a sucesivas generaciones Mutación SOMÁTICA: Afecta linea no germinal. Capacidad para originar clones (cels troncales). No hereditarias. Pueden originar tumores.
. No hereditarias. Pueden originar tumores.")
109
Examples of Point Mutations
Figure 3. Examples of Point Mutations. Panel A shows the normal sequence of DNA from one exon and the protein product it encodes. Panel B shows a silent mutation, Panel C a conservative missense mutation (serine and threonine have very similar structures), Panel D a nonconservative missense mutation (serine and proline have very different structures), Panel E a nonsense mutation, and Panel F a frame-shift mutation. In Panel F, the insertion of a single G throws off the reading frame, so that all amino acids downstream are changed radically. Guttmacher, A. E. et al. N Engl J Med 2002;347:
, Panel D a nonconservative missense mutation (serine and proline have very different structures), Panel E a nonsense mutation, and Panel F a frame-shift mutation. In Panel F, the insertion of a single G throws off the reading frame, so that all amino acids downstream are changed radically. Guttmacher, A. E. et al. N Engl J Med 2002;347:")
110
TIPOS DE MUTACIONES MUTACIÓN ESPONTANEA MUTACIÓN INDUCIDA
Condiciones ambientales normales MUTACIÓN INDUCIDA Condiciones ambientales específicas AGENTE MUTAGÉNICO Acelerador tasa espontanea mutación. Tasa: para un gen determinado y por generación (20 años)
")
111
PROTECCIÓN CONTRA MUTACIÓN
REPARACIÓN CÓDIGO GENÉTICO 61 CODONES; 20 AA; Alteraciones silentes POSICIÓN EN PROTEÍNA Zonas de proteínas no críticas para su función ERRORES AUTOSÓMICOS RECESIVOS Alelo funcional produce 50% proteína suficiente CONDICIONAL Déficit G6PD. Favismo. Primaquina.

112
PROTECCIÓN CONTRA MUTACIÓN
REPARACIÓN CÓDIGO GENÉTICO 61 CODONES; 20 AA; Alteraciones silentes POSICIÓN EN PROTEÍNA Zonas de proteínas no críticas para su función ERRORES AUTOSÓMICOS RECESIVOS Alelo funcional produce 50% proteína suficiente CONDICIONAL Déficit G6PD. Favismo. Primaquina.

113
Examples of Point Mutations
Figure 3. Examples of Point Mutations. Panel A shows the normal sequence of DNA from one exon and the protein product it encodes. Panel B shows a silent mutation, Panel C a conservative missense mutation (serine and threonine have very similar structures), Panel D a nonconservative missense mutation (serine and proline have very different structures), Panel E a nonsense mutation, and Panel F a frame-shift mutation. In Panel F, the insertion of a single G throws off the reading frame, so that all amino acids downstream are changed radically. Guttmacher, A. E. et al. N Engl J Med 2002;347:
, Panel D a nonconservative missense mutation (serine and proline have very different structures), Panel E a nonsense mutation, and Panel F a frame-shift mutation. In Panel F, the insertion of a single G throws off the reading frame, so that all amino acids downstream are changed radically. Guttmacher, A. E. et al. N Engl J Med 2002;347:")
114
The Thyrotropin Receptor
Figure 1. The Thyrotropin Receptor. The location of constitutively activating mutations1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16 and inactivating mutations15,17,18 of the thyrotropin-receptor gene is shown, as is the location of somatic mutations found in thyroid carcinomas.10,19,20 At some locations, several different amino acid substitutions have been described. All gain-of-function mutations are in exon 10 except Ser281Asn/Thr, which is in exon 9. Gain-of-function mutations are denoted by circles in the case of hyperfunctioning thyroid adenomas, squares in the case of familial autosomal dominant hyperthyroidism, diamonds in the case of sporadic congenital hyperthyroidism, and octagons in the case of thyroid carcinomas. Loss-of-function mutations are denoted by triangles. Letters indicate the amino acid in the wild-type receptor. The asterisk and double asterisk indicate deletions resulting in a gain of function in hyperfunctioning thyroid adenomas. Paschke, R. et al. N Engl J Med 1997;337:

115
Components of Myocyte Cytoarchitecture (Panel A) and Mutations Causing Dilated Cardiomyopathy and Conduction-System Disease or Autosomal Dominant Emery-Dreifuss Muscular Dystrophy (Panel B) Figure 1. Components of Myocyte Cytoarchitecture (Panel A) and Mutations Causing Dilated Cardiomyopathy and Conduction-System Disease or Autosomal Dominant Emery-Dreifuss Muscular Dystrophy (Panel B). Mutations in the rod domain of the lamin A/C gene cause isolated dilated cardiomyopathy and conduction-system disease, presumably through perturbed interactions with nuclear or cytoplasmic constituents (Panel A). Other cytoskeletal molecules implicated in the pathophysiology of human dilated cardiomyopathy include actin, dystrophin, and the dystrophin-associated glycoprotein complex.12,23,24,25,26,27,28 Interactions between lamins A and C and cytoskeletal or sarcomere proteins are unknown. Conduction-system disease is a common feature of Emery-Dreifuss muscular dystrophy caused by defects in the head or tail domain of the lamin gene or by emerin mutations. Mutations causing dilated cardiomyopathy and conduction-system disease or autosomal dominant Emery-Dreifuss muscular dystrophy are distributed in distinct domains of the lamin dimer (Panel B). Lamins A and C have identical structures throughout the amino-terminal head (NH3), {alpha}-helical rod domain, and proximal carboxyl-terminal tail (COOH), but they differ in their distal amino acids (lamin A is shown in gray, and lamin C is shown in black). Mutations in the rod domain (Arg60Gly, Leu85Arg, Asn195Lys, and Glu203Gly) cause dilated cardiomyopathy and conduction-system disease without skeletal myopathy; the mutation at the carboxyl terminal (Arg571Ser) is associated with subclinical skeletal-muscle disease. Mutations that cause Emery-Dreifuss muscular dystrophy (Gln6Stop, Arg453Trp, Arg527Pro, and Leu530Pro) do not affect the {alpha}-helical rod domain. Fatkin, D. et al. N Engl Med 1999;341: J
 and Mutations Causing Dilated Cardiomyopathy and Conduction-System Disease or Autosomal Dominant Emery-Dreifuss Muscular Dystrophy (Panel B). Mutations in the rod domain of the lamin A/C gene cause isolated dilated cardiomyopathy and conduction-system disease, presumably through perturbed interactions with nuclear or cytoplasmic constituents (Panel A). Other cytoskeletal molecules implicated in the pathophysiology of human dilated cardiomyopathy include actin, dystrophin, and the dystrophin-associated glycoprotein complex.12,23,24,25,26,27,28 Interactions between lamins A and C and cytoskeletal or sarcomere proteins are unknown. Conduction-system disease is a common feature of Emery-Dreifuss muscular dystrophy caused by defects in the head or tail domain of the lamin gene or by emerin mutations. Mutations causing dilated cardiomyopathy and conduction-system disease or autosomal dominant Emery-Dreifuss muscular dystrophy are distributed in distinct domains of the lamin dimer (Panel B). Lamins A and C have identical structures throughout the amino-terminal head (NH3), {alpha}-helical rod domain, and proximal carboxyl-terminal tail (COOH), but they differ in their distal amino acids (lamin A is shown in gray, and lamin C is shown in black). Mutations in the rod domain (Arg60Gly, Leu85Arg, Asn195Lys, and Glu203Gly) cause dilated cardiomyopathy and conduction-system disease without skeletal myopathy; the mutation at the carboxyl terminal (Arg571Ser) is associated with subclinical skeletal-muscle disease. Mutations that cause Emery-Dreifuss muscular dystrophy (Gln6Stop, Arg453Trp, Arg527Pro, and Leu530Pro) do not affect the {alpha}-helical rod domain. Fatkin, D. et al. N Engl Med 1999;341: J.")
116
PROTECCIÓN CONTRA MUTACIÓN
REPARACIÓN CÓDIGO GENÉTICO 61 CODONES; 20 AA; Alteraciones silentes POSICIÓN EN PROTEÍNA Zonas de proteínas no críticas para su función ERRORES AUTOSÓMICOS RECESIVOS Alelo funcional produce 50 proteína suficiente CONDICIONAL Déficit G6PD. Favismo

117
PROTECCIÓN CONTRA MUTACIÓN
REPARACIÓN CÓDIGO GENÉTICO 61 CODONES; 20 AA; Alteraciones silentes POSICIÓN EN PROTEÍNA Zonas de proteínas no críticas para su función ERRORES AUTOSÓMICOS RECESIVOS Alelo funcional produce 50% proteína suficiente CONDICIONAL Déficit G6PD. Favismo. Primaquina.

118
PROTECCIÓN CONTRA MUTACIÓN
REPARACIÓN Reconocimiento bases anormales GLICOSILASA: corta unión Base-Dribosa ENDONUCLEASA: reconoce lugar sin base y corta ADN POLIMERASA REPARADORA: introduce la base que falta. CÓDIGO GENÉTICO POSICIÓN EN PROTEÍNA ERRORES AUTOSÓMICOS RECESIVOS CONDICIONAL

119
The Life and Death of a Cisplatin Adduct
The Life and Death of a Cisplatin Adduct. In Panel A, a cisplatin molecule binds covalently to genomic DNA, forming a bulky, helix-distorting adduct. The most prevalent adduct is the intrastrand linkage of two adjacent guanine bases by the nitrogen atoms at position 7 (the GG adduct). In chemosensitive cells with low nucleotide excision repair activity, apoptosis usually follows. In chemoresistant cells with high nucleotide excision repair activity, the adduct may be excised and the DNA repaired. First, the adduct is recognized, and proteins of the nucleotide excision repair complex are assembled at the adduct site (Panel B). The heterodimeric protein excision repair cross-complementation group 1 (ERCC1)-XPF is the last component to be assembled -- the rate-limiting step. Unwinding of the DNA duplex in the immediate vicinity of the adduct results in the formation of a bubble. Next, endonucleases create dual incisions flanking the damaged bases (Panel C), with the protein XPG acting on the 3' side and the heterodimer ERCC1-XPF acting on the 5' side. The segment of about 22 to 32 nucleotides containing the adduct is removed. Then, the excised segment is repaired by polymerases and the accessory replication proteins PCNA, RPA, and RFC (Panel D). The integrity of the damaged strand is restored by DNA ligase. The protein ribonucleotide reductase M1 (RRM1), although not an integral part of the repair complex, catalyzes the biosynthesis of deoxyribonucleotides from the corresponding ribonucleotides, providing the building blocks for reconstitution of the excised oligonucleotide. The repair process is complete (Panel E), and the original state of the DNA is restored. (Modified from Friedberg.1) N Engl J Med 2007;356: Gazdar A.
. In chemosensitive cells with low nucleotide excision repair activity, apoptosis usually follows. In chemoresistant cells with high nucleotide excision repair activity, the adduct may be excised and the DNA repaired. First, the adduct is recognized, and proteins of the nucleotide excision repair complex are assembled at the adduct site (Panel B). The heterodimeric protein excision repair cross-complementation group 1 (ERCC1)-XPF is the last component to be assembled -- the rate-limiting step. Unwinding of the DNA duplex in the immediate vicinity of the adduct results in the formation of a bubble. Next, endonucleases create dual incisions flanking the damaged bases (Panel C), with the protein XPG acting on the 3 side and the heterodimer ERCC1-XPF acting on the 5 side. The segment of about 22 to 32 nucleotides containing the adduct is removed. Then, the excised segment is repaired by polymerases and the accessory replication proteins PCNA, RPA, and RFC (Panel D). The integrity of the damaged strand is restored by DNA ligase. The protein ribonucleotide reductase M1 (RRM1), although not an integral part of the repair complex, catalyzes the biosynthesis of deoxyribonucleotides from the corresponding ribonucleotides, providing the building blocks for reconstitution of the excised oligonucleotide. The repair process is complete (Panel E), and the original state of the DNA is restored. (Modified from Friedberg.1) N Engl J Med 2007;356: Gazdar A.")
120
The Life and Death of a Cisplatin Adduct
The Life and Death of a Cisplatin Adduct. In Panel A, a cisplatin molecule binds covalently to genomic DNA, forming a bulky, helix-distorting adduct. The most prevalent adduct is the intrastrand linkage of two adjacent guanine bases by the nitrogen atoms at position 7 (the GG adduct). In chemosensitive cells with low nucleotide excision repair activity, apoptosis usually follows. In chemoresistant cells with high nucleotide excision repair activity, the adduct may be excised and the DNA repaired. First, the adduct is recognized, and proteins of the nucleotide excision repair complex are assembled at the adduct site (Panel B). The heterodimeric protein excision repair cross-complementation group 1 (ERCC1)-XPF is the last component to be assembled -- the rate-limiting step. Unwinding of the DNA duplex in the immediate vicinity of the adduct results in the formation of a bubble. Next, endonucleases create dual incisions flanking the damaged bases (Panel C), with the protein XPG acting on the 3' side and the heterodimer ERCC1-XPF acting on the 5' side. The segment of about 22 to 32 nucleotides containing the adduct is removed. Then, the excised segment is repaired by polymerases and the accessory replication proteins PCNA, RPA, and RFC (Panel D). The integrity of the damaged strand is restored by DNA ligase. The protein ribonucleotide reductase M1 (RRM1), although not an integral part of the repair complex, catalyzes the biosynthesis of deoxyribonucleotides from the corresponding ribonucleotides, providing the building blocks for reconstitution of the excised oligonucleotide. The repair process is complete (Panel E), and the original state of the DNA is restored. (Modified from Friedberg.1) Gazdar A. N Engl J Med 2007;356:
. In chemosensitive cells with low nucleotide excision repair activity, apoptosis usually follows. In chemoresistant cells with high nucleotide excision repair activity, the adduct may be excised and the DNA repaired. First, the adduct is recognized, and proteins of the nucleotide excision repair complex are assembled at the adduct site (Panel B). The heterodimeric protein excision repair cross-complementation group 1 (ERCC1)-XPF is the last component to be assembled -- the rate-limiting step. Unwinding of the DNA duplex in the immediate vicinity of the adduct results in the formation of a bubble. Next, endonucleases create dual incisions flanking the damaged bases (Panel C), with the protein XPG acting on the 3 side and the heterodimer ERCC1-XPF acting on the 5 side. The segment of about 22 to 32 nucleotides containing the adduct is removed. Then, the excised segment is repaired by polymerases and the accessory replication proteins PCNA, RPA, and RFC (Panel D). The integrity of the damaged strand is restored by DNA ligase. The protein ribonucleotide reductase M1 (RRM1), although not an integral part of the repair complex, catalyzes the biosynthesis of deoxyribonucleotides from the corresponding ribonucleotides, providing the building blocks for reconstitution of the excised oligonucleotide. The repair process is complete (Panel E), and the original state of the DNA is restored. (Modified from Friedberg.1) Gazdar A. N Engl J Med 2007;356:")
121
The Life and Death of a Cisplatin Adduct
The Life and Death of a Cisplatin Adduct. In Panel A, a cisplatin molecule binds covalently to genomic DNA, forming a bulky, helix-distorting adduct. The most prevalent adduct is the intrastrand linkage of two adjacent guanine bases by the nitrogen atoms at position 7 (the GG adduct). In chemosensitive cells with low nucleotide excision repair activity, apoptosis usually follows. In chemoresistant cells with high nucleotide excision repair activity, the adduct may be excised and the DNA repaired. First, the adduct is recognized, and proteins of the nucleotide excision repair complex are assembled at the adduct site (Panel B). The heterodimeric protein excision repair cross-complementation group 1 (ERCC1)-XPF is the last component to be assembled -- the rate-limiting step. Unwinding of the DNA duplex in the immediate vicinity of the adduct results in the formation of a bubble. Next, endonucleases create dual incisions flanking the damaged bases (Panel C), with the protein XPG acting on the 3' side and the heterodimer ERCC1-XPF acting on the 5' side. The segment of about 22 to 32 nucleotides containing the adduct is removed. Then, the excised segment is repaired by polymerases and the accessory replication proteins PCNA, RPA, and RFC (Panel D). The integrity of the damaged strand is restored by DNA ligase. The protein ribonucleotide reductase M1 (RRM1), although not an integral part of the repair complex, catalyzes the biosynthesis of deoxyribonucleotides from the corresponding ribonucleotides, providing the building blocks for reconstitution of the excised oligonucleotide. The repair process is complete (Panel E), and the original state of the DNA is restored. (Modified from Friedberg.1) Gazdar A. N Engl J Med 2007;356:
. In chemosensitive cells with low nucleotide excision repair activity, apoptosis usually follows. In chemoresistant cells with high nucleotide excision repair activity, the adduct may be excised and the DNA repaired. First, the adduct is recognized, and proteins of the nucleotide excision repair complex are assembled at the adduct site (Panel B). The heterodimeric protein excision repair cross-complementation group 1 (ERCC1)-XPF is the last component to be assembled -- the rate-limiting step. Unwinding of the DNA duplex in the immediate vicinity of the adduct results in the formation of a bubble. Next, endonucleases create dual incisions flanking the damaged bases (Panel C), with the protein XPG acting on the 3 side and the heterodimer ERCC1-XPF acting on the 5 side. The segment of about 22 to 32 nucleotides containing the adduct is removed. Then, the excised segment is repaired by polymerases and the accessory replication proteins PCNA, RPA, and RFC (Panel D). The integrity of the damaged strand is restored by DNA ligase. The protein ribonucleotide reductase M1 (RRM1), although not an integral part of the repair complex, catalyzes the biosynthesis of deoxyribonucleotides from the corresponding ribonucleotides, providing the building blocks for reconstitution of the excised oligonucleotide. The repair process is complete (Panel E), and the original state of the DNA is restored. (Modified from Friedberg.1) Gazdar A. N Engl J Med 2007;356:")
122
The Life and Death of a Cisplatin Adduct
The Life and Death of a Cisplatin Adduct. In Panel A, a cisplatin molecule binds covalently to genomic DNA, forming a bulky, helix-distorting adduct. The most prevalent adduct is the intrastrand linkage of two adjacent guanine bases by the nitrogen atoms at position 7 (the GG adduct). In chemosensitive cells with low nucleotide excision repair activity, apoptosis usually follows. In chemoresistant cells with high nucleotide excision repair activity, the adduct may be excised and the DNA repaired. First, the adduct is recognized, and proteins of the nucleotide excision repair complex are assembled at the adduct site (Panel B). The heterodimeric protein excision repair cross-complementation group 1 (ERCC1)-XPF is the last component to be assembled -- the rate-limiting step. Unwinding of the DNA duplex in the immediate vicinity of the adduct results in the formation of a bubble. Next, endonucleases create dual incisions flanking the damaged bases (Panel C), with the protein XPG acting on the 3' side and the heterodimer ERCC1-XPF acting on the 5' side. The segment of about 22 to 32 nucleotides containing the adduct is removed. Then, the excised segment is repaired by polymerases and the accessory replication proteins PCNA, RPA, and RFC (Panel D). The integrity of the damaged strand is restored by DNA ligase. The protein ribonucleotide reductase M1 (RRM1), although not an integral part of the repair complex, catalyzes the biosynthesis of deoxyribonucleotides from the corresponding ribonucleotides, providing the building blocks for reconstitution of the excised oligonucleotide. The repair process is complete (Panel E), and the original state of the DNA is restored. (Modified from Friedberg.1) Gazdar A. N Engl J Med 2007;356:
. In chemosensitive cells with low nucleotide excision repair activity, apoptosis usually follows. In chemoresistant cells with high nucleotide excision repair activity, the adduct may be excised and the DNA repaired. First, the adduct is recognized, and proteins of the nucleotide excision repair complex are assembled at the adduct site (Panel B). The heterodimeric protein excision repair cross-complementation group 1 (ERCC1)-XPF is the last component to be assembled -- the rate-limiting step. Unwinding of the DNA duplex in the immediate vicinity of the adduct results in the formation of a bubble. Next, endonucleases create dual incisions flanking the damaged bases (Panel C), with the protein XPG acting on the 3 side and the heterodimer ERCC1-XPF acting on the 5 side. The segment of about 22 to 32 nucleotides containing the adduct is removed. Then, the excised segment is repaired by polymerases and the accessory replication proteins PCNA, RPA, and RFC (Panel D). The integrity of the damaged strand is restored by DNA ligase. The protein ribonucleotide reductase M1 (RRM1), although not an integral part of the repair complex, catalyzes the biosynthesis of deoxyribonucleotides from the corresponding ribonucleotides, providing the building blocks for reconstitution of the excised oligonucleotide. The repair process is complete (Panel E), and the original state of the DNA is restored. (Modified from Friedberg.1) Gazdar A. N Engl J Med 2007;356:")
123
The Life and Death of a Cisplatin Adduct
The Life and Death of a Cisplatin Adduct. In Panel A, a cisplatin molecule binds covalently to genomic DNA, forming a bulky, helix-distorting adduct. The most prevalent adduct is the intrastrand linkage of two adjacent guanine bases by the nitrogen atoms at position 7 (the GG adduct). In chemosensitive cells with low nucleotide excision repair activity, apoptosis usually follows. In chemoresistant cells with high nucleotide excision repair activity, the adduct may be excised and the DNA repaired. First, the adduct is recognized, and proteins of the nucleotide excision repair complex are assembled at the adduct site (Panel B). The heterodimeric protein excision repair cross-complementation group 1 (ERCC1)-XPF is the last component to be assembled -- the rate-limiting step. Unwinding of the DNA duplex in the immediate vicinity of the adduct results in the formation of a bubble. Next, endonucleases create dual incisions flanking the damaged bases (Panel C), with the protein XPG acting on the 3' side and the heterodimer ERCC1-XPF acting on the 5' side. The segment of about 22 to 32 nucleotides containing the adduct is removed. Then, the excised segment is repaired by polymerases and the accessory replication proteins PCNA, RPA, and RFC (Panel D). The integrity of the damaged strand is restored by DNA ligase. The protein ribonucleotide reductase M1 (RRM1), although not an integral part of the repair complex, catalyzes the biosynthesis of deoxyribonucleotides from the corresponding ribonucleotides, providing the building blocks for reconstitution of the excised oligonucleotide. The repair process is complete (Panel E), and the original state of the DNA is restored. (Modified from Friedberg.1) Gazdar A. N Engl J Med 2007;356:
. In chemosensitive cells with low nucleotide excision repair activity, apoptosis usually follows. In chemoresistant cells with high nucleotide excision repair activity, the adduct may be excised and the DNA repaired. First, the adduct is recognized, and proteins of the nucleotide excision repair complex are assembled at the adduct site (Panel B). The heterodimeric protein excision repair cross-complementation group 1 (ERCC1)-XPF is the last component to be assembled -- the rate-limiting step. Unwinding of the DNA duplex in the immediate vicinity of the adduct results in the formation of a bubble. Next, endonucleases create dual incisions flanking the damaged bases (Panel C), with the protein XPG acting on the 3 side and the heterodimer ERCC1-XPF acting on the 5 side. The segment of about 22 to 32 nucleotides containing the adduct is removed. Then, the excised segment is repaired by polymerases and the accessory replication proteins PCNA, RPA, and RFC (Panel D). The integrity of the damaged strand is restored by DNA ligase. The protein ribonucleotide reductase M1 (RRM1), although not an integral part of the repair complex, catalyzes the biosynthesis of deoxyribonucleotides from the corresponding ribonucleotides, providing the building blocks for reconstitution of the excised oligonucleotide. The repair process is complete (Panel E), and the original state of the DNA is restored. (Modified from Friedberg.1) Gazdar A. N Engl J Med 2007;356:")
124
The Life and Death of a Cisplatin Adduct
The Life and Death of a Cisplatin Adduct. In Panel A, a cisplatin molecule binds covalently to genomic DNA, forming a bulky, helix-distorting adduct. The most prevalent adduct is the intrastrand linkage of two adjacent guanine bases by the nitrogen atoms at position 7 (the GG adduct). In chemosensitive cells with low nucleotide excision repair activity, apoptosis usually follows. In chemoresistant cells with high nucleotide excision repair activity, the adduct may be excised and the DNA repaired. First, the adduct is recognized, and proteins of the nucleotide excision repair complex are assembled at the adduct site (Panel B). The heterodimeric protein excision repair cross-complementation group 1 (ERCC1)-XPF is the last component to be assembled -- the rate-limiting step. Unwinding of the DNA duplex in the immediate vicinity of the adduct results in the formation of a bubble. Next, endonucleases create dual incisions flanking the damaged bases (Panel C), with the protein XPG acting on the 3' side and the heterodimer ERCC1-XPF acting on the 5' side. The segment of about 22 to 32 nucleotides containing the adduct is removed. Then, the excised segment is repaired by polymerases and the accessory replication proteins PCNA, RPA, and RFC (Panel D). The integrity of the damaged strand is restored by DNA ligase. The protein ribonucleotide reductase M1 (RRM1), although not an integral part of the repair complex, catalyzes the biosynthesis of deoxyribonucleotides from the corresponding ribonucleotides, providing the building blocks for reconstitution of the excised oligonucleotide. The repair process is complete (Panel E), and the original state of the DNA is restored. (Modified from Friedberg.1) Gazdar A. N Engl J Med 2007;356:
. In chemosensitive cells with low nucleotide excision repair activity, apoptosis usually follows. In chemoresistant cells with high nucleotide excision repair activity, the adduct may be excised and the DNA repaired. First, the adduct is recognized, and proteins of the nucleotide excision repair complex are assembled at the adduct site (Panel B). The heterodimeric protein excision repair cross-complementation group 1 (ERCC1)-XPF is the last component to be assembled -- the rate-limiting step. Unwinding of the DNA duplex in the immediate vicinity of the adduct results in the formation of a bubble. Next, endonucleases create dual incisions flanking the damaged bases (Panel C), with the protein XPG acting on the 3 side and the heterodimer ERCC1-XPF acting on the 5 side. The segment of about 22 to 32 nucleotides containing the adduct is removed. Then, the excised segment is repaired by polymerases and the accessory replication proteins PCNA, RPA, and RFC (Panel D). The integrity of the damaged strand is restored by DNA ligase. The protein ribonucleotide reductase M1 (RRM1), although not an integral part of the repair complex, catalyzes the biosynthesis of deoxyribonucleotides from the corresponding ribonucleotides, providing the building blocks for reconstitution of the excised oligonucleotide. The repair process is complete (Panel E), and the original state of the DNA is restored. (Modified from Friedberg.1) Gazdar A. N Engl J Med 2007;356:")
125
PROTECCIÓN CONTRA MUTACIÓN
REPARACIÓN Reconocimiento bases anormales GLICOSILASA: corta unión Base-Dribosa ENDONUCLEASA: reconoce lugar sin base y corta ADN POLIMERASA REPARADORA: introduce la base que falta. DEFECTOS EN LA REPARACIÓN: Ataxia-telangiectasia, Anemia Fanconi, Xeroderma pigmentoso, Ca colon no polipósico.

126
La complejidad creciente del dogma central de la biología molecular
The Increasing Complexity of the Central Dogma of Molecular Biology. La complejidad creciente del dogma central de la biología molecular Figure 1. The Increasing Complexity of the Central Dogma of Molecular Biology. The flow of genomic information from DNA to RNA to protein remains the basis for understanding genomic function (Panel A). A single gene can yield an extensive array of gene products, depending on the environment in which it is expressed, thereby expanding the repertoire of the 20,000 or so genes in the human genome (Panel B). The initial event of gene expression, transcription, is regulated by means of a complex choreography of events involving the three-dimensional DNA structure, covalent chemical, or epigenetic, modifications of the DNA backbone, and interactions between protein and DNA and between RNA and DNA. Translation is similarly complex and tightly regulated by interactions between messenger RNA (mRNA) and proteins. Processing of single-precursor RNA (preRNA) molecules can yield multiple RNA products, including microRNA (miRNA) and small interfering RNA (siRNA) molecules. Post-translational modification of proteins also contributes greatly to the diversity of the output of the human genome through modifications of individual immature proteins (e.g., folding, cleavage, and chemical modifications), which yield an array of related protein products. Feero WG et al. N Engl J Med 2010;362:
. A single gene can yield an extensive array of gene products, depending on the environment in which it is expressed, thereby expanding the repertoire of the 20,000 or so genes in the human genome (Panel B). The initial event of gene expression, transcription, is regulated by means of a complex choreography of events involving the three-dimensional DNA structure, covalent chemical, or epigenetic, modifications of the DNA backbone, and interactions between protein and DNA and between RNA and DNA. Translation is similarly complex and tightly regulated by interactions between messenger RNA (mRNA) and proteins. Processing of single-precursor RNA (preRNA) molecules can yield multiple RNA products, including microRNA (miRNA) and small interfering RNA (siRNA) molecules. Post-translational modification of proteins also contributes greatly to the diversity of the output of the human genome through modifications of individual immature proteins (e.g., folding, cleavage, and chemical modifications), which yield an array of related protein products. Feero WG et al. N Engl J Med 2010;362:")
127
The Increasing Complexity of the Central Dogma of Molecular Biology.
Figure 1. The Increasing Complexity of the Central Dogma of Molecular Biology. The flow of genomic information from DNA to RNA to protein remains the basis for understanding genomic function (Panel A). A single gene can yield an extensive array of gene products, depending on the environment in which it is expressed, thereby expanding the repertoire of the 20,000 or so genes in the human genome (Panel B). The initial event of gene expression, transcription, is regulated by means of a complex choreography of events involving the three-dimensional DNA structure, covalent chemical, or epigenetic, modifications of the DNA backbone, and interactions between protein and DNA and between RNA and DNA. Translation is similarly complex and tightly regulated by interactions between messenger RNA (mRNA) and proteins. Processing of single-precursor RNA (preRNA) molecules can yield multiple RNA products, including microRNA (miRNA) and small interfering RNA (siRNA) molecules. Post-translational modification of proteins also contributes greatly to the diversity of the output of the human genome through modifications of individual immature proteins (e.g., folding, cleavage, and chemical modifications), which yield an array of related protein products. Feero WG et al. N Engl J Med 2010;362:
. A single gene can yield an extensive array of gene products, depending on the environment in which it is expressed, thereby expanding the repertoire of the 20,000 or so genes in the human genome (Panel B). The initial event of gene expression, transcription, is regulated by means of a complex choreography of events involving the three-dimensional DNA structure, covalent chemical, or epigenetic, modifications of the DNA backbone, and interactions between protein and DNA and between RNA and DNA. Translation is similarly complex and tightly regulated by interactions between messenger RNA (mRNA) and proteins. Processing of single-precursor RNA (preRNA) molecules can yield multiple RNA products, including microRNA (miRNA) and small interfering RNA (siRNA) molecules. Post-translational modification of proteins also contributes greatly to the diversity of the output of the human genome through modifications of individual immature proteins (e.g., folding, cleavage, and chemical modifications), which yield an array of related protein products. Feero WG et al. N Engl J Med 2010;362:")
128
MicroRNA No codifican proteínas.
Su producto es una fibra simple de RNA de 21 a 23 nucleótidos. Función: regular la expresión de genes. Puede bloquear la traslación de un RNA mensajero o degradarlo. Puede ser supresores si su diana es un oncogen o se pueden comportar como oncogenes si su diana es un gen supresor Pueden activarse (amplificación) o inactivarse (delección, silenciado epigenético o pérdida de expresión de factores de transcripción)
 o inactivarse (delección, silenciado epigenético o pérdida de expresión de factores de transcripción)")
129
Cambios epigenéticos Cambios heredables en la estructura de un gen sin alterar la secuencia del DNA. Los dos más frecuentes son la METILACIÓN y la modificación de las histonas por acetilación. METILACIÓN: Unión de grupo metilo al carbono 5 en una isla CpG (región de 500 pares de bases con alto contenido en Citosina y Guanina). Hipermetilación : silenciamiento de genes. Hipometilación: nivel de transcripción elevado
. Hipermetilación : silenciamiento de genes. Hipometilación: nivel de transcripción elevado.")
130
Genética en Patología General II
Remigio Cordero Torres Badajoz 14 de Septiembre 2015

131
FENOTIPO = GENOTIPO + AMBIENTE
Resultado de la constitución genetica heredada más los factores ambientales Enfermedad: Resultado de la interacción entre la constitución genética y el entorno

132
ENFERMEDADES GENÉTICAS
MONOGÉNICAS MENDELIANAS Herencia AUTOSÓMICA DOMINANTE Herencia AUTOSÓMICA RECESIVA Herencia LIGADA AL SEXO (ligada a X) MULTIFACTORIAL Frecuente Poligénica a veces monogénica Importante componente ambiental CROMOSOMOPATÍAS ALTERACIONES NUMÉRICAS ALTERACIONES ESTRUCTURALES
 MULTIFACTORIAL. Frecuente Poligénica a veces monogénica. Importante componente ambiental. CROMOSOMOPATÍAS. ALTERACIONES NUMÉRICAS. ALTERACIONES ESTRUCTURALES.")
133
ENFERMEDADES GENÉTICAS
MONOGÉNICAS MENDELIANAS Herencia AUTOSÓMICA DOMINANTE Herencia AUTOSÓMICA RECESIVA Herencia LIGADA AL SEXO (ligada a X) MULTIFACTORIAL Frecuente Poligénica a veces monogénica Importante componente ambiental CROMOSOMOPATÍAS ALTERACIONES NUMÉRICAS ALTERACIONES ESTRUCTURALES
 MULTIFACTORIAL. Frecuente Poligénica a veces monogénica. Importante componente ambiental. CROMOSOMOPATÍAS. ALTERACIONES NUMÉRICAS. ALTERACIONES ESTRUCTURALES.")
134
Los genes en el individuo diploide se encuentran por parejas ocupando cada uno un lugar o locus equivalente en cada cromosoma homólogo. Si el gen que ocupa un locus es idéntico al que ocupa ese locus en el cromosoma homólogo el individuo es homocigoto para ese gen. Si es distinto es heterocigoto. Los heterocigotos pueden expresar una forma del gen, las dos formas o una forma intermedia o mezclada. Las variantes de un gen se denominan alelos

135
PENETRANCIA Capacidad que tiene una alelo mutado dominante para expresarse en el fenotipo de un heterocigótico Penetrancia de 80%: El 80% de individuos con el alelo mutado tienen manifestaciones clínicas

136
EXPRESIVIDAD Capacidad de un alelo mutado dominante para desarrollar en un individuo todas o parte de las manifestaciones clínicas.

137
EXPRESIVIDAD Capacidad de un alelo mutado dominante para desarrollar en un individuo todas o parte de las manifestaciones clínicas. Penetrancia: tiene o no tiene polidactilia Expresividad; Polidactilia en uno, dos, tres o todos los miembros

138
HERENCIA AUTOSÓMICA DOMINANTE
Alelo se expresa en heterocigotos REGLAS Arbol genealógico transmisión vertical Varones y hembras afectados por igual Afectados en genealogías grandes tiende a 50% (riesgo de transmitir a hijo/a) Cada enfermo tiene un progenitor afectado Penetrancia reducida y expresividad variable
 Cada enfermo tiene un progenitor afectado. Penetrancia reducida y expresividad variable.")
139
Autosomal Dominant Inheritance
Figure 1. Autosomal Dominant Inheritance. Children (indicated by the arrows) whose parent is affected by multiple endocrine neoplasia type 2 (MEN-2) have a 50 percent chance of inheriting the condition. Testing can identify the disease in such persons before clinical complications occur. Prophylactic thyroidectomy can be offered to those at risk, to prevent medullary thyroid carcinoma. Squares denote male family members, and circles female family members. Burke, W. N Engl J Med 2002;347:
 whose parent is affected by multiple endocrine neoplasia type 2 (MEN-2) have a 50 percent chance of inheriting the condition. Testing can identify the disease in such persons before clinical complications occur. Prophylactic thyroidectomy can be offered to those at risk, to prevent medullary thyroid carcinoma. Squares denote male family members, and circles female family members. Burke, W. N Engl J Med 2002;347:")
140
50. ¿Cuál crees que es la probabilidad de que un varón portador de una enfermedad autosómica dominante transmita su enfermedad a sus tres hijos?

141
50. ¿Cuál crees que es la probabilidad de que un varón portador de una enfermedad autosómica dominante transmita su enfermedad a sus tres hijos? a) 50% b) 25% c) 12.5% d) 100% e) 1%
 50% b) 25% c) 12.5% d) 100% e) 1%")
142
Cuando diagnosticando al feto diagnosticamos al padre.
Pedro descubre a los 24 años que su madre de 45 años, tiene corea de Huntington. Ella fue adoptada, por lo que no sabemos su historia familiar. La mujer de Pedro está embarazada y no quiere tener un hijo que tendrá enfermedad de Huntington. Quiere que se estudie si el feto es portador de la mutación. Pero Pedro no quiere conocer su posible estado de portador.

143
HERENCIA AUTOSÓMICA RECESIVA
Alelo que se expresa en homocigotos REGLAS Arboles genealógicos transmisión horizontal Varones y hembras afectados por igual Los dos padres de un afectado son portadores Riesgo de hijo afectado de padre y madre portadores: 25% Muy poca variabilidad fenotípica Frecuente consanguinidad entre padres de afectado

144
Autosomal Recessive Inheritance
Figure 3. Autosomal Recessive Inheritance. When an autosomal recessive condition such as sickle cell anemia is diagnosed in a child (indicated by the arrow), the parents are identified as carriers of the sickle cell trait, which is inherited. All children of these parents have a 25 percent chance of being affected. Children who do not have sickle cell anemia have a 67 percent chance of being carriers. Cystic fibrosis is also inherited as an autosomal recessive condition. Burke, W. N Engl J Med 2002;347:
, the parents are identified as carriers of the sickle cell trait, which is inherited. All children of these parents have a 25 percent chance of being affected. Children who do not have sickle cell anemia have a 67 percent chance of being carriers. Cystic fibrosis is also inherited as an autosomal recessive condition. Burke, W. N Engl J Med 2002;347:")
145
Mucociliary Transport in the Healthy Lung and in Cystic Fibrosis.
Figure 1. Mucociliary Transport in the Healthy Lung and in Cystic Fibrosis. A fluid layer that is maintained through a balance of chloride secretion and sodium absorption covers the surface of airway epithelial cells. The airway surface fluid supports a thin mucus layer produced by mucosal secretory glands. The mucus layer is transported by respiratory cilia from the lower airways to the airway opening (Panel A). In cystic fibrosis, defective chloride secretion and sodium hyperabsorption lead to the depletion of the layer of airway surface fluid, with consecutive breakdown of mucociliary transport (Panel B). Osmotic agents increase the volume of airway surface fluid through an increase in water influx, thereby restoring mucociliary function (Panel C). For simplicity, inflammatory cells are not shown. Ratjen F. N Engl J Med 2006;354:
. In cystic fibrosis, defective chloride secretion and sodium hyperabsorption lead to the depletion of the layer of airway surface fluid, with consecutive breakdown of mucociliary transport (Panel B). Osmotic agents increase the volume of airway surface fluid through an increase in water influx, thereby restoring mucociliary function (Panel C). For simplicity, inflammatory cells are not shown. Ratjen F. N Engl J Med 2006;354:")
146
Mucociliary Transport in the Healthy Lung and in Cystic Fibrosis.
Ratjen F. N Engl J Med 2006; 354: Figure 1. Mucociliary Transport in the Healthy Lung and in Cystic Fibrosis. A fluid layer that is maintained through a balance of chloride secretion and sodium absorption covers the surface of airway epithelial cells. The airway surface fluid supports a thin mucus layer produced by mucosal secretory glands. The mucus layer is transported by respiratory cilia from the lower airways to the airway opening (Panel A). In cystic fibrosis, defective chloride secretion and sodium hyperabsorption lead to the depletion of the layer of airway surface fluid, with consecutive breakdown of mucociliary transport (Panel B). Osmotic agents increase the volume of airway surface fluid through an increase in water influx, thereby restoring mucociliary function (Panel C). For simplicity, inflammatory cells are not shown.
. In cystic fibrosis, defective chloride secretion and sodium hyperabsorption lead to the depletion of the layer of airway surface fluid, with consecutive breakdown of mucociliary transport (Panel B). Osmotic agents increase the volume of airway surface fluid through an increase in water influx, thereby restoring mucociliary function (Panel C). For simplicity, inflammatory cells are not shown.")
147
FIBROSIS QUÍSTICA Patogénesis enfermedad pulmonar
Mutación gen RTFQ (CFTR) cr7-> Proteína RTFQ anormal (proteína de canal de cloro) Retención de iones y agua Secreción mucosa anormal Acumulación de secreción Hipoventilación pulmonar Inflamación mucosa. Enfermedad pulmonar crónica
 cr7-> Proteína RTFQ anormal (proteína de canal de cloro) Retención de iones y agua. Secreción mucosa anormal. Acumulación de secreción. Hipoventilación pulmonar. Inflamación mucosa. Enfermedad pulmonar crónica.")
148
HERENCIA LIGADA AL SEXO RECESIVA
Varones afectados y hembras portadoras REGLAS - Arbol genealógico en “salto de caballo” - Hembra portadora con 50% hijos afectados 50% hijas portadoras - Varón afectado: 100% hijos normales 100% hijas portadoras - No hay transmisión varón-varón.

149
X-Linked Recessive Inheritance
Figure 2. X-Linked Recessive Inheritance. A woman (indicated by the arrow) wants to know whether she carries the gene for Duchenne's muscular dystrophy (DMD), because her uncle and her brother were both affected (solid symbols), and her mother and grandmother are known to be carriers (symbols with a solid center). She has a 50 percent chance of inheriting the carrier status from her mother. Genetic testing can be used to determine her carrier status if her affected brother has a positive test result. Squares denote male family members, and circles female family members. Burke, W. N Engl J Med 2002;347:
 wants to know whether she carries the gene for Duchenne s muscular dystrophy (DMD), because her uncle and her brother were both affected (solid symbols), and her mother and grandmother are known to be carriers (symbols with a solid center). She has a 50 percent chance of inheriting the carrier status from her mother. Genetic testing can be used to determine her carrier status if her affected brother has a positive test result. Squares denote male family members, and circles female family members. Burke, W. N Engl J Med 2002;347:")
150
Mechanism of PRO051 in the Restoration of Dystrophin Expression through Exon Skipping.
Figure 1. Mechanism of PRO051 in the Restoration of Dystrophin Expression through Exon Skipping. Normal muscle produces dystrophin, a critical protein, in response to signals encoded by the enormous DMD gene, in which 79 exons are spliced together in a precise lockstep manner into messenger RNA (mRNA). The mRNA is then translated into dystrophin protein. All exons are spliced to maintain the triplet codon reading frame required for effective protein translation (Panel A). In the muscle of patients with Duchenne's muscular dystrophy, mutations in the dystrophin gene lead to the loss of one or more exons (Panel B, in which exon 50 is deleted). The mRNA splices together the remaining exons; however, the triplet reading frame is not maintained, which leads to errors in translation (frame shift) and loss of production of the dystrophin protein. The loss of dystrophin at the plasma membrane leads to the secondary loss of other associated membrane cytoskeleton structures. This, in turn, leads to membrane fragility, an abnormal influx of calcium ions (Ca 2+), and the efflux of creatine kinase (CK) into the patient's blood. Intramuscular injection of the small modified DNA molecule called PRO051 probably enters the Duchenne's muscle through abnormal muscle membranes; it then enters the nucleus and binds to the dystrophin mRNA in a sequence-specific manner (Panel C). In hybridization to the mRNA, PRO051 blocks the splicing machinery and prevents the inclusion of an additional exon (exon 51 in this example). The skipping of this additional exon restores the reading frame of the mRNA, allowing new production of dystrophin. The dystrophin that is produced is not normal but probably retains considerable function, as evidenced by the condition of patients with clinically milder Becker's muscular dystrophy who have similar or identically modified dystrophins. Hoffman EP. N Engl J Med 2007;357:
. The mRNA is then translated into dystrophin protein. All exons are spliced to maintain the triplet codon reading frame required for effective protein translation (Panel A). In the muscle of patients with Duchenne s muscular dystrophy, mutations in the dystrophin gene lead to the loss of one or more exons (Panel B, in which exon 50 is deleted). The mRNA splices together the remaining exons; however, the triplet reading frame is not maintained, which leads to errors in translation (frame shift) and loss of production of the dystrophin protein. The loss of dystrophin at the plasma membrane leads to the secondary loss of other associated membrane cytoskeleton structures. This, in turn, leads to membrane fragility, an abnormal influx of calcium ions (Ca 2+), and the efflux of creatine kinase (CK) into the patient s blood. Intramuscular injection of the small modified DNA molecule called PRO051 probably enters the Duchenne s muscle through abnormal muscle membranes; it then enters the nucleus and binds to the dystrophin mRNA in a sequence-specific manner (Panel C). In hybridization to the mRNA, PRO051 blocks the splicing machinery and prevents the inclusion of an additional exon (exon 51 in this example). The skipping of this additional exon restores the reading frame of the mRNA, allowing new production of dystrophin. The dystrophin that is produced is not normal but probably retains considerable function, as evidenced by the condition of patients with clinically milder Becker s muscular dystrophy who have similar or identically modified dystrophins. Hoffman EP. N Engl J Med 2007;357:")
151
HERENCIA LIGADA AL SEXO DOMINANTE
Varones y mujeres afectadas REGLAS - Hembra afectada: 50% hijos afectados 50% hijas afectadas - Varón afectado: 100% hijos normales 100% hijas afectadas - No hay transmisión varón-varón.

152
HERENCIA MITOCONDRIAL
Mitocondrias: varias copias de “minicromosoma” que contiene 37 genes. Alta Producción radic libres + no enzimas reparadoras: más mutaciones. La madre transmite todas sus mitocondrias Afectados hijos e hijas de mujer afectada Síntomas en tejidos con muchas mitocondrias: músculo esquelético: fatiga. Atrofia óptica de Leber MELAS (miopatía, mitocondrias, encefalopatía, ácido lactico, síndrome)
")
153
Mitochondrial DNA (mtDNA) and the Mitochondrial Respiratory Chain.
Figure 2. Mitochondrial DNA (mtDNA) and the Mitochondrial Respiratory Chain. Panel A shows the map of the human mitochondrial genome. The protein-coding genes — seven subunits of complex I (ND), three subunits of cytochrome c oxidase ( COX), the cytochrome b subunit of complex III ( Cyt b), and two subunits of adenosine triphosphate (ATP) synthase ( A6 and A8) — are shown in red. The protein-synthesis genes — the 12S and 16S ribosomal RNAs and the 22 transfer RNAs (three-letter amino acid symbols) — are shown in blue. The D-loop region controls the initiation of replication and transcription of mtDNA. Panel B shows the subunits of the respiratory chain encoded by nuclear DNA (nDNA) in blue and the subunits encoded by mtDNA in red. As electrons (e –) flow along the electron-transport chain, protons (H +) are pumped from the matrix to the intermembrane space through complexes I, III, and IV and then back into the matrix through complex V, to produce ATP. Coenzyme Q (CoQ) and cytochrome c (Cyt c) are electron-transfer carriers. Genes responsible for the indicated respiratory-chain disorders are also shown. ATPase 6 denotes ATP synthase 6; BCS1L cytochrome b–c complex assembly protein(complex III); NDUF NADH dehydrogenase–ubiquinone oxidoreductase; SCO synthesis of cytochrome oxidase; SDHA, SDHB, SDHC, and SDHD succinate dehydrogenase subunits; SURF1 surfeit gene 1; FBSN familial bilateral striatal necrosis; LHON Leber's hereditary optic neuropathy; MELAS mitochondrial encephalomyopathy, lactic acidosis, and strokelike episodes; MILS maternally inherited Leigh's syndrome; NARP neuropathy, ataxia, and retinitis pigmentosa; GRACILE growth retardation, aminoaciduria, lactic acidosis, and early death; and ALS amyotrophic lateral sclerosis. DiMauro S, Schon EA. N Engl J Med 2003;348:
 and the Mitochondrial Respiratory Chain. Panel A shows the map of the human mitochondrial genome. The protein-coding genes — seven subunits of complex I (ND), three subunits of cytochrome c oxidase ( COX), the cytochrome b subunit of complex III ( Cyt b), and two subunits of adenosine triphosphate (ATP) synthase ( A6 and A8) — are shown in red. The protein-synthesis genes — the 12S and 16S ribosomal RNAs and the 22 transfer RNAs (three-letter amino acid symbols) — are shown in blue. The D-loop region controls the initiation of replication and transcription of mtDNA. Panel B shows the subunits of the respiratory chain encoded by nuclear DNA (nDNA) in blue and the subunits encoded by mtDNA in red. As electrons (e –) flow along the electron-transport chain, protons (H +) are pumped from the matrix to the intermembrane space through complexes I, III, and IV and then back into the matrix through complex V, to produce ATP. Coenzyme Q (CoQ) and cytochrome c (Cyt c) are electron-transfer carriers. Genes responsible for the indicated respiratory-chain disorders are also shown. ATPase 6 denotes ATP synthase 6; BCS1L cytochrome b–c complex assembly protein(complex III); NDUF NADH dehydrogenase–ubiquinone oxidoreductase; SCO synthesis of cytochrome oxidase; SDHA, SDHB, SDHC, and SDHD succinate dehydrogenase subunits; SURF1 surfeit gene 1; FBSN familial bilateral striatal necrosis; LHON Leber s hereditary optic neuropathy; MELAS mitochondrial encephalomyopathy, lactic acidosis, and strokelike episodes; MILS maternally inherited Leigh s syndrome; NARP neuropathy, ataxia, and retinitis pigmentosa; GRACILE growth retardation, aminoaciduria, lactic acidosis, and early death; and ALS amyotrophic lateral sclerosis. DiMauro S, Schon EA. N Engl J Med 2003;348:")
154
Mutations in the Human Mitochondrial Genome That Are Known to Cause Disease.
Figure 3. Mutations in the Human Mitochondrial Genome That Are Known to Cause Disease. Disorders that are frequently or prominently associated with mutations in a particular gene are shown in boldface. Diseases due to mutations that impair mitochondrial protein synthesis are shown in blue. Diseases due to mutations in protein-coding genes are shown in red. ECM denotes encephalomyopathy; FBSN familial bilateral striatal necrosis; LHON Leber's hereditary optic neuropathy; LS Leigh's syndrome; MELAS mitochondrial encephalomyopathy, lactic acidosis, and strokelike episodes; MERRF myoclonic epilepsy with ragged-red fibers; MILS maternally inherited Leigh's syndrome; NARP neuropathy, ataxia, and retinitis pigmentosa; PEO progressive external ophthalmoplegia; PPK palmoplantar keratoderma; and SIDS sudden infant death syndrome. DiMauro S, Schon EA. N Engl J Med 2003;348:

155
Serial Cross-Sections of Muscle from a Patient with the Kearns–Sayre Syndrome, Showing Increased Mitochondrial Activity in Ragged-Red Fibers on Staining with Succinate Dehydrogenase (Asterisks in Panel A; ×120) and the Absence of Activity on Staining with Cytochrome c Oxidase (Asterisks in Panel B; ×120). Figure 4. Serial Cross-Sections of Muscle from a Patient with the Kearns–Sayre Syndrome, Showing Increased Mitochondrial Activity in Ragged-Red Fibers on Staining with Succinate Dehydrogenase (Asterisks in Panel A; ×120) and the Absence of Activity on Staining with Cytochrome c Oxidase (Asterisks in Panel B; ×120). DiMauro S, Schon EA. N Engl J Med 2003;348:
 and the Absence of Activity on Staining with Cytochrome c Oxidase (Asterisks in Panel B; ×120). DiMauro S, Schon EA. N Engl J Med 2003;348:")
156
ENFERMEDADES GENÉTICAS
MONOGÉNICAS MENDELIANAS Herencia AUTOSÓMICA DOMINANTE Herencia AUTOSÓMICA RECESIVA Herencia LIGADA AL SEXO (ligada a X) MULTIFACTORIAL Frecuente Poligénica a veces monogénica Importante componente ambiental CROMOSOMOPATÍAS ALTERACIONES NUMÉRICAS ALTERACIONES ESTRUCTURALES
 MULTIFACTORIAL. Frecuente Poligénica a veces monogénica. Importante componente ambiental. CROMOSOMOPATÍAS. ALTERACIONES NUMÉRICAS. ALTERACIONES ESTRUCTURALES.")
157
ENFERMEDADES GENÉTICAS
MONOGÉNICAS MENDELIANAS Herencia AUTOSÓMICA DOMINANTE Herencia AUTOSÓMICA RECESIVA Herencia LIGADA AL SEXO (ligada a X) MULTIFACTORIAL Frecuente Poligénica a veces monogénica Importante componente ambiental CROMOSOMOPATÍAS ALTERACIONES NUMÉRICAS ALTERACIONES ESTRUCTURALES
 MULTIFACTORIAL. Frecuente Poligénica a veces monogénica. Importante componente ambiental. CROMOSOMOPATÍAS. ALTERACIONES NUMÉRICAS. ALTERACIONES ESTRUCTURALES.")
158
Concordancia entre gemelos
Enfermedad Monocigóticos Dicigóticos Acné 14% Alzheimer 78% 39% Anorexia nerviosa 55% 7% Autismo 90% 4.5% Desorden Bipolar 33-80% 0-8% Labio leporino 40% 3-6% Hipertensión arterial 62% 48% Esquizofrenia 40-50% 10%

159
ENFERMEDADES GENÉTICAS
MONOGÉNICAS MENDELIANAS Herencia AUTOSÓMICA DOMINANTE Herencia AUTOSÓMICA RECESIVA Herencia LIGADA AL SEXO (ligada a X) MULTIFACTORIAL Frecuente Poligénica a veces monogénica Importante componente ambiental CROMOSOMOPATÍAS ALTERACIONES NUMÉRICAS ALTERACIONES ESTRUCTURALES
 MULTIFACTORIAL. Frecuente Poligénica a veces monogénica. Importante componente ambiental. CROMOSOMOPATÍAS. ALTERACIONES NUMÉRICAS. ALTERACIONES ESTRUCTURALES.")
160
Enfermedades GENÉTICAS (II)
CROMOSOMOPATÍAS ALTERACIONES NUMÉRICAS Autosómicas; Trisomías 21(Down), 18 (Edwards), 13(Patau) Monosomías (letales). Mosaicos: 47/46 Sexuales: Trisomía XXY(Klinefelter), XXX, XYY(Jacobs) Monosomía XO (Turner) Polisomías XXXY, XXXXY, XXXX Mosaicos XXY/XY; XO/XX ALTERACIONES ESTRUCTURALES Deleciones, Duplicaciones, Traslocaciones, Inversiones, Isocromosomas, Anulares.
, 18 (Edwards), 13(Patau) Monosomías (letales). Mosaicos: 47/46. Sexuales: Trisomía XXY(Klinefelter), XXX, XYY(Jacobs) Monosomía XO (Turner) Polisomías XXXY, XXXXY, XXXX. Mosaicos XXY/XY; XO/XX. ALTERACIONES ESTRUCTURALES. Deleciones, Duplicaciones, Traslocaciones, Inversiones, Isocromosomas, Anulares.")
161
Fluorescence in Situ Hybridization Showing the 22q11 Microdeletion Syndrome
Figure 4. Fluorescence in Situ Hybridization Showing the 22q11 Microdeletion Syndrome. An orange probe identifies the chromosomal segment that is deleted in the syndrome; thus, the chromosome 22 with the microdeletion -- del(22) -- lacks this probe. A green probe identifies a different segment of the chromosome and is used as a marker for the two copies of chromosome 22, one of which is normal and thus demonstrates both probes (22). Photomicrograph provided courtesy of Dr. Christine Disteche and Douglas Chapman, University of Washington. Burke, W. N Engl J Med 2002;347:
 -- lacks this probe. A green probe identifies a different segment of the chromosome and is used as a marker for the two copies of chromosome 22, one of which is normal and thus demonstrates both probes (22). Photomicrograph provided courtesy of Dr. Christine Disteche and Douglas Chapman, University of Washington. Burke, W. N Engl J Med 2002;347:")
162
Características de las CROMOSOMOPATÍAS
1. Se detectan con MICROSCOPIO ÓPTICO. 2. Se originan frecuentemente durante la MEIOSIS de uno de los progenitores 3. Enormes BLOQUES de genes: NUMEROSOS y graves síntomas 4. NO SON HEREDABLES

163
Características anomalías más frecuentes
Síndrome Cariotipo Género Frecuencia de nacimientos Síntomas Turner 45X Hembra 1/2500 Baja estatura Infertilidad Cuello palmeado Anomalías renales Trisomía X 47 XXX 1/1000 Talla alta Rasgos faciales anómalos Hipotonía Klinefelter 47 XXY Varón Lomgilineo Déficit lenguaje

164
Genes asociados a Trisomía 21 (Sdr Down)
Producto de Gen Signos y síntomas (Fenotipo) Proteína precursor de Amiloide (APP) Deoósito de proteínas en cerebro Factor 1 de ensamblaje proteína (CAF1A) Síntesis de DNA detenida Colágeno tipo VI (COL6A1) Defectos cardiacos Cristalino (CRYA1) Cataratas Receptor de Interferon (IFNAR) Immunidad enlentecida Kinasa I (DYRK1A) Retardo mental Oncoproteína ETS2 (ETS2) Anormalidades esqueléticas, cancer Fosforibosil glicil amina glicilTr (GART) Fallo en síntesis y reparación de DNA Superoxido dismutasa (SOD) Envejecimiento prematuro
 Proteína precursor de Amiloide (APP) Deoósito de proteínas en cerebro. Factor 1 de ensamblaje proteína (CAF1A) Síntesis de DNA detenida. Colágeno tipo VI (COL6A1) Defectos cardiacos. Cristalino (CRYA1) Cataratas. Receptor de Interferon (IFNAR) Immunidad enlentecida. Kinasa I (DYRK1A) Retardo mental. Oncoproteína ETS2 (ETS2) Anormalidades esqueléticas, cancer. Fosforibosil glicil amina glicilTr (GART) Fallo en síntesis y reparación de DNA. Superoxido dismutasa (SOD) Envejecimiento prematuro.")
165
- manipulación de la dieta sobre AMBIENTE: - ingeniería ambiental
POSIBILIDADES DE ACTUACIÓN sobre FENOTIPO: - manipulación de la dieta - transplante de órganos sobre AMBIENTE: - ingeniería ambiental - políticas sociales, educativas, sanitarias sobre GENOTIPO:

166
El retraso de Sanidad bloquea la selección genética de hijos para salvar a un hermano El País, domingo 1 de Octubre de 2006 1. Niño con enfermedad incurable 2. Fecundación in vitro 3. Analisis de compatibilidad de embriones 4. Implante de embrión compatible 5. Nace un niño sano que puede hacer de donante 6. Trasplante

167
Robert Edwards. Premio Nobel de Medicina 2010 por sus investigaciones sobre la fecundación in vitro.

168
Premio Nobel de Medicina 9 Octubre 2007
Mario R Capecci, Martin J Evans, Oliver Smithies Base teórica y experimental para la modificación genética dirigida en el ratón Martin Evans en 1981 obtiene cultivos de células pruripotentes embrionarias Mario Capecci y Oliver Smithies desarrollan métodos para modificar estas células de forma controlada inactivando genes específicos. Los ratones obtenidos a partir de estas células geneticamente modificadas manifestaban los efectos de la mutación de un gen determinado.

169
Terapia Génica 2003: China. Transferencia de p53 para tumores d cabeza y cuello. 2008: Argentina. Inserción de un gen que dificulta metabolización de alcohol ingerido. 2008: EEUU, RU. Tratamiento de amaurosis congénita de Leber (Ceguera antes de 30 años) inyectanto virus con gen RPE65. 2009: Aiuti. Milán.Tratamiento inmunodeficiencia combinada severa por déficit de ADA.
 inyectanto virus con gen RPE : Aiuti. Milán.Tratamiento inmunodeficiencia combinada severa por déficit de ADA.")
172
Sandhaus RA. N Engl J Med 2012;366:567-569.
Ameliorating Alpha1-Antitrypsin Deficiency. Figure 1. Ameliorating Alpha1-Antitrypsin Deficiency. This diagram presents a model of how a man or woman with alpha1-antitrypsin deficiency might be treated with autologous corrected induced pluripotent stem cells (iPSCs), as described by Yusa et al. 1 If such corrective therapy is successful, gene-corrected hepatocytes derived from the patient's own iPSCs would populate and perhaps replace the existing liver cells. As damaged native hepatocytes die consequent to an excess of mutant alpha1-antitrypsin protein, abnormal hepatocytes might be replaced by corrected iPSC-derived hepatocytes. Sandhaus RA. N Engl J Med 2012;366:
, as described by Yusa et al. 1 If such corrective therapy is successful, gene-corrected hepatocytes derived from the patient s own iPSCs would populate and perhaps replace the existing liver cells. As damaged native hepatocytes die consequent to an excess of mutant alpha1-antitrypsin protein, abnormal hepatocytes might be replaced by corrected iPSC-derived hepatocytes. Sandhaus RA. N Engl J Med 2012;366:")
173
Terapia génica basada en RNAi (RNA interferencia)
Las células han desarrollado un sistema para detectar cuando les invade un RNA extraño (infecciones virales) reconocerlo y evitar que se genere la proteína correspondiente. RNA de interferencia artificiales pueden apagar cualquier gen, de un organismo invasor o del propio genoma. RNAi molécula del año para Science en Premio Nobel 2006: Andrew Fire y Craig C Mello. microRNA: fabricado por nuestro propio genoma para interferir la expresíón de nuestros genes. Enero 2008: Massagué micro RNA evitan MTS
 reconocerlo y evitar que se genere la proteína correspondiente. RNA de interferencia artificiales pueden apagar cualquier gen, de un organismo invasor o del propio genoma. RNAi molécula del año para Science en Premio Nobel 2006: Andrew Fire y Craig C Mello. microRNA: fabricado por nuestro propio genoma para interferir la expresíón de nuestros genes. Enero 2008: Massagué micro RNA evitan MTS.")
174
Protocolos de terapia génica aprobados y publicados (Febrero 2013)
Enfermedad Objetivo Déficit de ADA Reemplazamiento de ADA Déficit Alfa-1 antitripsina Reemplazamiento alfa 1- antitripsina AIDS Inactivación antígeno presentación HIV Cáncer Refuerzo función inmune Quimioprotección Ablación tumoral Fibrosis quística Reemplazamiento enzima reguladora Hipercolesterolemia familiar Reemplazamiento receptores LDL Anemia de Fanconi Liberación Gen grupo C Complemento Enfermedad de Gaucher Reemplazamiento Glococerebridasa Hemofilia B Reemplazamiento Factor IX Artritis reumatoide Liberación Citokinas

175
BASES GENÉTICAS DEL CANCER
Patología General 15 de Septiembre de 2015 Remigio Cordero Torres

176
Singular Discoveries and Major Events in the Cancer Field and Changing Relative Survival Rates for Patients with Cancer in the United States, 1863–2006. DeVita VT Jr, Rosenberg SA . N Engl J Med 2012;366:

177
CÁNCER: pérdida de control del ciclo celular genéticamente determinada
NO ES UNA ENFERMEDAD HEREDITARIA PUEDE EXISTIR PREDISPOSICIÓN CONSTITUCIONAL PUEDE CONSIDERARSE UNA ENFERMEDAD GENÉTICA

178
CÁNCER: pérdida de control del ciclo celular genéticamente determinada
NO ES UNA ENFERMEDAD HEREDITARIA PUEDE EXISTIR PREDISPOSICIÓN CONSTITUCIONAL PUEDE CONSIDERARSE UNA ENFERMEDAD GENÉTICA

179
CONTROL DEL CICLO CELULAR
RELOJ CELULAR: SEÑALES EXTERNAS EFECTO MULTITUD (Crowded) HORMONAS FACTORES DE CRECIMIENTO SEÑALES INTERNAS CICLINAS Y KINASAS:
 HORMONAS. FACTORES DE CRECIMIENTO. SEÑALES INTERNAS. CICLINAS Y KINASAS:")
180
Figure 1. Mutations in Genes That Regulate Cellular Proliferation and the Repair of DNA and Lead to the Development of Breast Cancer. The ATM (ataxia-telangiectasia mutated) gene encodes a kinase that phosphorylates and thus activates the tumor-suppressor p53 either directly or by activating CHK2 in response to damage to DNA. The p53 protein then triggers the arrest of the cell cycle, allowing time for DNA to be repaired. The BRCA1 and NBS proteins are also activated by ATM and are thought to be directly involved in the repair of damaged DNA. The inactivating mutations in the genes that encode these proteins and thus lead to or increase the risk of breast cancer are shown. G1 and G2 are gap 1 and 2. Haber, D. N Engl J Med 2000;343:
 gene encodes a kinase that phosphorylates and thus activates the tumor-suppressor p53 either directly or by activating CHK2 in response to damage to DNA. The p53 protein then triggers the arrest of the cell cycle, allowing time for DNA to be repaired. The BRCA1 and NBS proteins are also activated by ATM and are thought to be directly involved in the repair of damaged DNA. The inactivating mutations in the genes that encode these proteins and thus lead to or increase the risk of breast cancer are shown. G1 and G2 are gap 1 and 2. Haber, D. N Engl J Med 2000;343:")
181
RELOJ CELULAR Cels tejido conectivo FETO en cultivo : 50 divisiones Cels tejido conectivo ADULTO en cultivo: divisiones

182
Figure 1. Telomere Structure
Figure 1. Telomere Structure. As shown in Panel A, telomeres are located at the ends of linear chromosomes; they are composed of hundreds to thousands of tandem DNA repeat sequences: hexameric TTAGGG in the leading strand and CCCTAA in the lagging strand in humans. Protective proteins associated with telomere DNA are collectively termed shelterin (TRF1, TRF2, TIN2, POT1, TPP1, and RAP1). The 3′ end of the telomeric leading strand terminates as a single-stranded overhang, which folds back and invades the double-stranded telomeric helix, forming the T loop. As shown in Panel B, telomeres can be directly visualized under the microscope at the ends of metaphase chromosomes (four telomere signals per chromosome) by fluorescence in situ hybridization (FISH). (Image provided by Peter Lansdorp, M.D., Ph.D.) Average telomere length can be measured by several methods: a technique that combines flow cytometry and FISH (flow-FISH), Southern blotting, and a quantitative polymerase-chain-reaction (qPCR) assay. Flow-FISH can measure the telomere length in different cell subgroups, such as granulocytes or CD4+ T lymphocytes; Southern blotting reveals length and length heterogeneity; and qPCR is a rapid assay that requires very small amounts of DNA. As shown in Panel C, the average length of telomeres in human leukocytes varies, ranging from approximately 11 kb at birth (in umbilical-cord blood) to 6 kb at 90 years of age. Telomere loss is most rapid early in life, and over a life span it is not linear but follows a third-order polynomial. Data are from Yamaguchi et al.6 Telomere Structure. Calado RT, Young NS. N Engl J Med 2009;361:
. The 3′ end of the telomeric leading strand terminates as a single-stranded overhang, which folds back and invades the double-stranded telomeric helix, forming the T loop. As shown in Panel B, telomeres can be directly visualized under the microscope at the ends of metaphase chromosomes (four telomere signals per chromosome) by fluorescence in situ hybridization (FISH). (Image provided by Peter Lansdorp, M.D., Ph.D.) Average telomere length can be measured by several methods: a technique that combines flow cytometry and FISH (flow-FISH), Southern blotting, and a quantitative polymerase-chain-reaction (qPCR) assay. Flow-FISH can measure the telomere length in different cell subgroups, such as granulocytes or CD4+ T lymphocytes; Southern blotting reveals length and length heterogeneity; and qPCR is a rapid assay that requires very small amounts of DNA. As shown in Panel C, the average length of telomeres in human leukocytes varies, ranging from approximately 11 kb at birth (in umbilical-cord blood) to 6 kb at 90 years of age. Telomere loss is most rapid early in life, and over a life span it is not linear but follows a third-order polynomial. Data are from Yamaguchi et al.6. Telomere Structure. Calado RT, Young NS. N Engl J Med 2009;361:")
183
Figure 1. Telomere Structure
Figure 1. Telomere Structure. As shown in Panel A, telomeres are located at the ends of linear chromosomes; they are composed of hundreds to thousands of tandem DNA repeat sequences: hexameric TTAGGG in the leading strand and CCCTAA in the lagging strand in humans. Protective proteins associated with telomere DNA are collectively termed shelterin (TRF1, TRF2, TIN2, POT1, TPP1, and RAP1). The 3′ end of the telomeric leading strand terminates as a single-stranded overhang, which folds back and invades the double-stranded telomeric helix, forming the T loop. As shown in Panel B, telomeres can be directly visualized under the microscope at the ends of metaphase chromosomes (four telomere signals per chromosome) by fluorescence in situ hybridization (FISH). (Image provided by Peter Lansdorp, M.D., Ph.D.) Average telomere length can be measured by several methods: a technique that combines flow cytometry and FISH (flow-FISH), Southern blotting, and a quantitative polymerase-chain-reaction (qPCR) assay. Flow-FISH can measure the telomere length in different cell subgroups, such as granulocytes or CD4+ T lymphocytes; Southern blotting reveals length and length heterogeneity; and qPCR is a rapid assay that requires very small amounts of DNA. As shown in Panel C, the average length of telomeres in human leukocytes varies, ranging from approximately 11 kb at birth (in umbilical-cord blood) to 6 kb at 90 years of age. Telomere loss is most rapid early in life, and over a life span it is not linear but follows a third-order polynomial. Data are from Yamaguchi et al.6 Telomere Structure. Calado RT, Young NS. N Engl J Med 2009;361:
. The 3′ end of the telomeric leading strand terminates as a single-stranded overhang, which folds back and invades the double-stranded telomeric helix, forming the T loop. As shown in Panel B, telomeres can be directly visualized under the microscope at the ends of metaphase chromosomes (four telomere signals per chromosome) by fluorescence in situ hybridization (FISH). (Image provided by Peter Lansdorp, M.D., Ph.D.) Average telomere length can be measured by several methods: a technique that combines flow cytometry and FISH (flow-FISH), Southern blotting, and a quantitative polymerase-chain-reaction (qPCR) assay. Flow-FISH can measure the telomere length in different cell subgroups, such as granulocytes or CD4+ T lymphocytes; Southern blotting reveals length and length heterogeneity; and qPCR is a rapid assay that requires very small amounts of DNA. As shown in Panel C, the average length of telomeres in human leukocytes varies, ranging from approximately 11 kb at birth (in umbilical-cord blood) to 6 kb at 90 years of age. Telomere loss is most rapid early in life, and over a life span it is not linear but follows a third-order polynomial. Data are from Yamaguchi et al.6. Telomere Structure. Calado RT, Young NS. N Engl J Med 2009;361:")
184
Figure 1. Telomere Structure
Figure 1. Telomere Structure. As shown in Panel A, telomeres are located at the ends of linear chromosomes; they are composed of hundreds to thousands of tandem DNA repeat sequences: hexameric TTAGGG in the leading strand and CCCTAA in the lagging strand in humans. Protective proteins associated with telomere DNA are collectively termed shelterin (TRF1, TRF2, TIN2, POT1, TPP1, and RAP1). The 3′ end of the telomeric leading strand terminates as a single-stranded overhang, which folds back and invades the double-stranded telomeric helix, forming the T loop. As shown in Panel B, telomeres can be directly visualized under the microscope at the ends of metaphase chromosomes (four telomere signals per chromosome) by fluorescence in situ hybridization (FISH). (Image provided by Peter Lansdorp, M.D., Ph.D.) Average telomere length can be measured by several methods: a technique that combines flow cytometry and FISH (flow-FISH), Southern blotting, and a quantitative polymerase-chain-reaction (qPCR) assay. Flow-FISH can measure the telomere length in different cell subgroups, such as granulocytes or CD4+ T lymphocytes; Southern blotting reveals length and length heterogeneity; and qPCR is a rapid assay that requires very small amounts of DNA. As shown in Panel C, the average length of telomeres in human leukocytes varies, ranging from approximately 11 kb at birth (in umbilical-cord blood) to 6 kb at 90 years of age. Telomere loss is most rapid early in life, and over a life span it is not linear but follows a third-order polynomial. Data are from Yamaguchi et al.6 Telomere Structure. Calado RT, Young NS. N Engl J Med 2009;361:
. The 3′ end of the telomeric leading strand terminates as a single-stranded overhang, which folds back and invades the double-stranded telomeric helix, forming the T loop. As shown in Panel B, telomeres can be directly visualized under the microscope at the ends of metaphase chromosomes (four telomere signals per chromosome) by fluorescence in situ hybridization (FISH). (Image provided by Peter Lansdorp, M.D., Ph.D.) Average telomere length can be measured by several methods: a technique that combines flow cytometry and FISH (flow-FISH), Southern blotting, and a quantitative polymerase-chain-reaction (qPCR) assay. Flow-FISH can measure the telomere length in different cell subgroups, such as granulocytes or CD4+ T lymphocytes; Southern blotting reveals length and length heterogeneity; and qPCR is a rapid assay that requires very small amounts of DNA. As shown in Panel C, the average length of telomeres in human leukocytes varies, ranging from approximately 11 kb at birth (in umbilical-cord blood) to 6 kb at 90 years of age. Telomere loss is most rapid early in life, and over a life span it is not linear but follows a third-order polynomial. Data are from Yamaguchi et al.6. Telomere Structure. Calado RT, Young NS. N Engl J Med 2009;361:")
185
RELOJ CELULAR Cels tejido conectivo FETO en cultivo : 50 divisiones
Cels tejido conectivo ADULTO en cultivo: 15-30 divisiones TELÓMEROS: bases repiten secuencia TTAGGG En cada mitosis: pérdida de bases Después de aproximadamente 50 mitosis: STOP

186
Gametos normales y células de cáncer
Células somáticas normales

187
TELOMERASA

188
Structure and Function of Telomerase
Figure 1. Structure and Function of Telomerase. Telomerase comprises two subunits, telomerase reverse transcriptase (TERT) and the telomerase RNA component (TERC). Telomerase adds telomere repeats to the ends of chromosomes by reverse transcribing the sequence in the template region of TERC (boxed) from RNA to DNA in six-nucleotide increments (sequences shown in red). Telomerase can continue elongating telomeres through a ratcheting mechanism, by repeatedly dissociating from the newly synthesized telomere, realigning, and adding another six nucleotides at a time. Telomerase up-regulation is critical in cancer, allowing cancer cells to divide an unlimited number of times. Artandi S. N Engl J Med 2006;355:
 and the telomerase RNA component (TERC). Telomerase adds telomere repeats to the ends of chromosomes by reverse transcribing the sequence in the template region of TERC (boxed) from RNA to DNA in six-nucleotide increments (sequences shown in red). Telomerase can continue elongating telomeres through a ratcheting mechanism, by repeatedly dissociating from the newly synthesized telomere, realigning, and adding another six nucleotides at a time. Telomerase up-regulation is critical in cancer, allowing cancer cells to divide an unlimited number of times. Artandi S. N Engl J Med 2006;355:")
189
The Telomerase Complex and Its Components.
Figure 3. The Telomerase Complex and Its Components. The enzyme telomerase reverse transcriptase (TERT), its RNA component (TERC), the protein dyskerin, and other associated proteins (NHP2, NOP10, and GAR1) are shown. Telomerase catalytically adds TTAGGG hexameric nucleotide repeats to the 3′-hydroxyl end of the telomeric leading strand, using a specific sequence in the RNA component as the template. TERT contains three major domains: the N-terminal region, the reverse-transcriptase motifs, and the C-terminal region, all containing evolutionarily conserved motifs. TERC contains 451 nucleotides in seven conserved regions (CR1 through CR7), including the template (CR1), and an H/ACA box, a hairpin nucleotide sequence characteristic of a class of small nucleolar RNAs involved in RNA processing. Calado RT, Young NS. N Engl J Med 2009;361:
, its RNA component (TERC), the protein dyskerin, and other associated proteins (NHP2, NOP10, and GAR1) are shown. Telomerase catalytically adds TTAGGG hexameric nucleotide repeats to the 3′-hydroxyl end of the telomeric leading strand, using a specific sequence in the RNA component as the template. TERT contains three major domains: the N-terminal region, the reverse-transcriptase motifs, and the C-terminal region, all containing evolutionarily conserved motifs. TERC contains 451 nucleotides in seven conserved regions (CR1 through CR7), including the template (CR1), and an H/ACA box, a hairpin nucleotide sequence characteristic of a class of small nucleolar RNAs involved in RNA processing. Calado RT, Young NS. N Engl J Med 2009;361:")
190
RELOJ CELULAR Cels tejido conectivo FETO en cultivo : 50 divisiones Cels tejido conectivo ADULTO en cultivo: divisiones TELÓMEROS: bases repiten secuencia TTAGGG En cada mitosis: pérdida de bases Después de aproximadamente 50 mitosis: STOP TELOMERASA: (gametos, cels MO, Cáncer) añade nuevo DNA a telómeros-> continua división celular (inmortalidad celular)
 añade nuevo DNA a telómeros-> continua división celular (inmortalidad celular)")
191
CONTROL DEL CICLO CELULAR
RELOJ CELULAR: SEÑALES EXTERNAS EFECTO MULTITUD (Crowded) HORMONAS: FSH/LH -> UTERO: MITOSIS FACTORES DE CRECIMIENTO:locales Factor crecimiento epidérmico; estimula crecimiento tejido nervioso bajo una herida SEÑALES INTERNAS CICLINAS Y KINASAS: nivel de ciclinas aumenta en interfase y en cierto nivel se une a kinasas iniciando mitosis-> secreción de enzimas que degradan la ciclina
 HORMONAS: FSH/LH -> UTERO: MITOSIS. FACTORES DE CRECIMIENTO:locales. Factor crecimiento epidérmico; estimula crecimiento tejido nervioso bajo una herida. SEÑALES INTERNAS. CICLINAS Y KINASAS: nivel de ciclinas aumenta en interfase y en cierto nivel se une a kinasas iniciando mitosis-> secreción de enzimas que degradan la ciclina.")
192
CICLINA Figure 1. Mutations in Genes That Regulate Cellular Proliferation and the Repair of DNA and Lead to the Development of Breast Cancer. The ATM (ataxia-telangiectasia mutated) gene encodes a kinase that phosphorylates and thus activates the tumor-suppressor p53 either directly or by activating CHK2 in response to damage to DNA. The p53 protein then triggers the arrest of the cell cycle, allowing time for DNA to be repaired. The BRCA1 and NBS proteins are also activated by ATM and are thought to be directly involved in the repair of damaged DNA. The inactivating mutations in the genes that encode these proteins and thus lead to or increase the risk of breast cancer are shown. G1 and G2 are gap 1 and 2. Haber, D. N Engl J Med 2000;343:
 gene encodes a kinase that phosphorylates and thus activates the tumor-suppressor p53 either directly or by activating CHK2 in response to damage to DNA. The p53 protein then triggers the arrest of the cell cycle, allowing time for DNA to be repaired. The BRCA1 and NBS proteins are also activated by ATM and are thought to be directly involved in the repair of damaged DNA. The inactivating mutations in the genes that encode these proteins and thus lead to or increase the risk of breast cancer are shown. G1 and G2 are gap 1 and 2. Haber, D. N Engl J Med 2000;343:")
193
Cyclin D1 and the Development of Breast Cancer
Figure 1. Cyclin D1 and the Development of Breast Cancer. Cyclin D1 has a unique role in regulating the cell cycle in alveolar epithelial cells of the breast (Panel A). It is a possible therapeutic target because it is overexpressed in breast cancer, especially estrogen-receptor-positive breast cancers. Trastuzumab, the clinically useful monoclonal antibody against NEU, may indirectly inhibit cyclin D1 through the RAS pathway. In mice, the actions of the Neu and Ras oncogenes in the breast depend on cyclin D1, as suggested in Panel B. Mammary cancer induced in mice by overexpression of the Neu and Ras oncogenes depends on cyclin D1, whereas the Myc and Wnt1 oncogenes can act independently of cyclin D1. Chodosh, L. A. N Engl J Med 2002;347:
. It is a possible therapeutic target because it is overexpressed in breast cancer, especially estrogen-receptor-positive breast cancers. Trastuzumab, the clinically useful monoclonal antibody against NEU, may indirectly inhibit cyclin D1 through the RAS pathway. In mice, the actions of the Neu and Ras oncogenes in the breast depend on cyclin D1, as suggested in Panel B. Mammary cancer induced in mice by overexpression of the Neu and Ras oncogenes depends on cyclin D1, whereas the Myc and Wnt1 oncogenes can act independently of cyclin D1. Chodosh, L. A. N Engl J Med 2002;347:")
194
Cyclin D1 and the Development of Breast Cancer
Figure 1. Cyclin D1 and the Development of Breast Cancer. Cyclin D1 has a unique role in regulating the cell cycle in alveolar epithelial cells of the breast (Panel A). It is a possible therapeutic target because it is overexpressed in breast cancer, especially estrogen-receptor-positive breast cancers. Trastuzumab, the clinically useful monoclonal antibody against NEU, may indirectly inhibit cyclin D1 through the RAS pathway. In mice, the actions of the Neu and Ras oncogenes in the breast depend on cyclin D1, as suggested in Panel B. Mammary cancer induced in mice by overexpression of the Neu and Ras oncogenes depends on cyclin D1, whereas the Myc and Wnt1 oncogenes can act independently of cyclin D1. Chodosh, L. A. N Engl J Med 2002;347:
. It is a possible therapeutic target because it is overexpressed in breast cancer, especially estrogen-receptor-positive breast cancers. Trastuzumab, the clinically useful monoclonal antibody against NEU, may indirectly inhibit cyclin D1 through the RAS pathway. In mice, the actions of the Neu and Ras oncogenes in the breast depend on cyclin D1, as suggested in Panel B. Mammary cancer induced in mice by overexpression of the Neu and Ras oncogenes depends on cyclin D1, whereas the Myc and Wnt1 oncogenes can act independently of cyclin D1. Chodosh, L. A. N Engl J Med 2002;347:")
195
CÁNCER: pérdida de control del ciclo celular geneticamente determinada
NO ES UNA ENFERMEDAD HEREDITARIA PUEDE EXISTIR PREDISPOSICIÓN CONSTITUCIONAL PUEDE CONSIDERARSE UNA ENFERMEDAD GENÉTICA

196
CÁNCER: pérdida de control del ciclo celular geneticamente determinada
NO ES UNA ENFERMEDAD HEREDITARIA PUEDE EXISTIR PREDISPOSICIÓN CONSTITUCIONAL PUEDE CONSIDERARSE UNA ENFERMEDAD GENÉTICA

197
EL CÁNCER NO ES UNA ENFERMEDAD HEREDITARIA
Gemelos HOMOCIGÓTICOS baja concordancia neoplásica . Descendientes víctimas HIROSHIMA No aumento incidencia de leucemias . EMIGRACIÓN cambio patrón epidemiológico . Análisis CÁNCERES PROFESIONALES

198
CÁNCER: pérdida de control del ciclo celular geneticamente determinada
NO ES UNA ENFERMEDAD HEREDITARIA PUEDE EXISTIR PREDISPOSICIÓN CONSTITUCIONAL PUEDE CONSIDERARSE UNA ENFERMEDAD GENÉTICA

199
PUEDE EXISTIR PREDISPOSICIÓN CONSTITUCIONAL AL CÁNCER
SÍNDROMES INESTABILIDAD CROMOSÓMICA Ataxia telangiectasia. Sdr Bloom. A Fanconi. X pigmentosum CÁNCERES FAMILIARES Li Fraumeni. Retinoblastoma. Wilms. Ca Colon no polipósico ANOMALÍAS CONSTITUCIONALES Síndrome de Down-> Leucemias. XXY -> Neoplasias INMUNODEFICIENCIAS Variabilidad individual en el manejo de carcinógenos

200
PUEDE EXISTIR PREDISPOSICIÓN CONSTITUCIONAL AL CÁNCER
SÍNDROMES INESTABILIDAD CROMOSÓMICA Ataxia telangiectasia. Sdr Bloom. A Fanconi. X pigmentosum CÁNCERES FAMILIARES Li Fraumeni. Retinoblastoma. Wilms. Ca Colon no polipósico ANOMALÍAS CONSTITUCIONALES Síndrome de Down-> Leucemias. XXY -> Neoplasias INMUNODEFICIENCIAS Variabilidad individual en el manejo de carcinógenos

201
Singular Discoveries and Major Events in the Cancer Field and Changing Relative Survival Rates for Patients with Cancer in the United States, 1863–2006. DeVita VT Jr, Rosenberg SA . N Engl J Med 2012;366:

202
Initial (Panel A) and Subsequent (Panels B, C, and D) Evaluations of a Pedigree with Classic Hereditary Nonpolyposis Colorectal Cancer Figure 4. Initial (Panel A) and Subsequent (Panels B, C, and D) Evaluations of a Pedigree with Classic Hereditary Nonpolyposis Colorectal Cancer. Panel A shows the initial assessment of what turned out to be a family with classic hereditary nonpolyposis colorectal cancer (HNPCC). The proband (Subject IV-1; arrow) had early-onset (40 years) colorectal carcinoma and carcinoma of the ureter. These findings by themselves are highly significant clinically. However, his mother (Subject III-1) had uterine cervical carcinoma at the age of 37 years, a tumor not associated with the syndrome, but had colorectal carcinoma at the age of 55 years. In Panel B, further inquiry indicated that the proband's mother (Subject III-1) had two sisters with endometrial carcinoma at the ages of 57 (Subject III-2) and 61 (Subject III-3). This pattern, notwithstanding the Amsterdam criteria, would be sufficient for a diagnosis of hereditary nonpolyposis colorectal cancer. In Panel C, extending the pedigree further showed that one of these maternal aunts (Subject III-2) had three sons with cancer, one with early-onset metachronous colon cancer (Subject IV-3), a second with colon cancer and carcinoma of the ureter (Subject IV-4), and the third with colon cancer alone (Subject IV-5). The other aunt (Subject III-3) had a daughter (Subject IV-6) with cancer of the bile duct. These findings provide strong evidence in support of the diagnosis of hereditary nonpolyposis colorectal cancer. In Panel D, the full pedigree shows findings that continue to support a diagnosis of hereditary nonpolyposis colorectal cancer.55 Squares denote male family members; circles female family members; symbols with a slash deceased family members, with the age at death (d) given below each symbol; open symbols unaffected family members; bicolored symbols family members with multiple primary cancers; squares containing numbers the number of unaffected male progeny; circles containing numbers the number of unaffected female progeny; and combined symbols containing numbers the number of unaffected progeny of both sexes. The types of primary cancer and the age (in years) at diagnosis are listed below the symbols, and the bottom-most number is the current age or the age at death. Inf denotes in infancy. Lynch, H. T. et al. N Engl J Med 2003;348:
 and Subsequent (Panels B, C, and D) Evaluations of a Pedigree with Classic Hereditary Nonpolyposis Colorectal Cancer. Panel A shows the initial assessment of what turned out to be a family with classic hereditary nonpolyposis colorectal cancer (HNPCC). The proband (Subject IV-1; arrow) had early-onset (40 years) colorectal carcinoma and carcinoma of the ureter. These findings by themselves are highly significant clinically. However, his mother (Subject III-1) had uterine cervical carcinoma at the age of 37 years, a tumor not associated with the syndrome, but had colorectal carcinoma at the age of 55 years. In Panel B, further inquiry indicated that the proband s mother (Subject III-1) had two sisters with endometrial carcinoma at the ages of 57 (Subject III-2) and 61 (Subject III-3). This pattern, notwithstanding the Amsterdam criteria, would be sufficient for a diagnosis of hereditary nonpolyposis colorectal cancer. In Panel C, extending the pedigree further showed that one of these maternal aunts (Subject III-2) had three sons with cancer, one with early-onset metachronous colon cancer (Subject IV-3), a second with colon cancer and carcinoma of the ureter (Subject IV-4), and the third with colon cancer alone (Subject IV-5). The other aunt (Subject III-3) had a daughter (Subject IV-6) with cancer of the bile duct. These findings provide strong evidence in support of the diagnosis of hereditary nonpolyposis colorectal cancer. In Panel D, the full pedigree shows findings that continue to support a diagnosis of hereditary nonpolyposis colorectal cancer.55. Squares denote male family members; circles female family members; symbols with a slash deceased family members, with the age at death (d) given below each symbol; open symbols unaffected family members; bicolored symbols family members with multiple primary cancers; squares containing numbers the number of unaffected male progeny; circles containing numbers the number of unaffected female progeny; and combined symbols containing numbers the number of unaffected progeny of both sexes. The types of primary cancer and the age (in years) at diagnosis are listed below the symbols, and the bottom-most number is the current age or the age at death. Inf denotes in infancy. Lynch, H. T. et al. N Engl J Med 2003;348:")
203
Initial (Panel A) and Subsequent (Panels B, C, and D) Evaluations of a Pedigree with Classic Hereditary Nonpolyposis Colorectal Cancer Figure 4. Initial (Panel A) and Subsequent (Panels B, C, and D) Evaluations of a Pedigree with Classic Hereditary Nonpolyposis Colorectal Cancer. Panel A shows the initial assessment of what turned out to be a family with classic hereditary nonpolyposis colorectal cancer (HNPCC). The proband (Subject IV-1; arrow) had early-onset (40 years) colorectal carcinoma and carcinoma of the ureter. These findings by themselves are highly significant clinically. However, his mother (Subject III-1) had uterine cervical carcinoma at the age of 37 years, a tumor not associated with the syndrome, but had colorectal carcinoma at the age of 55 years. In Panel B, further inquiry indicated that the proband's mother (Subject III-1) had two sisters with endometrial carcinoma at the ages of 57 (Subject III-2) and 61 (Subject III-3). This pattern, notwithstanding the Amsterdam criteria, would be sufficient for a diagnosis of hereditary nonpolyposis colorectal cancer. In Panel C, extending the pedigree further showed that one of these maternal aunts (Subject III-2) had three sons with cancer, one with early-onset metachronous colon cancer (Subject IV-3), a second with colon cancer and carcinoma of the ureter (Subject IV-4), and the third with colon cancer alone (Subject IV-5). The other aunt (Subject III-3) had a daughter (Subject IV-6) with cancer of the bile duct. These findings provide strong evidence in support of the diagnosis of hereditary nonpolyposis colorectal cancer. In Panel D, the full pedigree shows findings that continue to support a diagnosis of hereditary nonpolyposis colorectal cancer.55 Squares denote male family members; circles female family members; symbols with a slash deceased family members, with the age at death (d) given below each symbol; open symbols unaffected family members; bicolored symbols family members with multiple primary cancers; squares containing numbers the number of unaffected male progeny; circles containing numbers the number of unaffected female progeny; and combined symbols containing numbers the number of unaffected progeny of both sexes. The types of primary cancer and the age (in years) at diagnosis are listed below the symbols, and the bottom-most number is the current age or the age at death. Inf denotes in infancy. Lynch, H. T. et al. N Engl J Med 2003;348:
 and Subsequent (Panels B, C, and D) Evaluations of a Pedigree with Classic Hereditary Nonpolyposis Colorectal Cancer. Panel A shows the initial assessment of what turned out to be a family with classic hereditary nonpolyposis colorectal cancer (HNPCC). The proband (Subject IV-1; arrow) had early-onset (40 years) colorectal carcinoma and carcinoma of the ureter. These findings by themselves are highly significant clinically. However, his mother (Subject III-1) had uterine cervical carcinoma at the age of 37 years, a tumor not associated with the syndrome, but had colorectal carcinoma at the age of 55 years. In Panel B, further inquiry indicated that the proband s mother (Subject III-1) had two sisters with endometrial carcinoma at the ages of 57 (Subject III-2) and 61 (Subject III-3). This pattern, notwithstanding the Amsterdam criteria, would be sufficient for a diagnosis of hereditary nonpolyposis colorectal cancer. In Panel C, extending the pedigree further showed that one of these maternal aunts (Subject III-2) had three sons with cancer, one with early-onset metachronous colon cancer (Subject IV-3), a second with colon cancer and carcinoma of the ureter (Subject IV-4), and the third with colon cancer alone (Subject IV-5). The other aunt (Subject III-3) had a daughter (Subject IV-6) with cancer of the bile duct. These findings provide strong evidence in support of the diagnosis of hereditary nonpolyposis colorectal cancer. In Panel D, the full pedigree shows findings that continue to support a diagnosis of hereditary nonpolyposis colorectal cancer.55. Squares denote male family members; circles female family members; symbols with a slash deceased family members, with the age at death (d) given below each symbol; open symbols unaffected family members; bicolored symbols family members with multiple primary cancers; squares containing numbers the number of unaffected male progeny; circles containing numbers the number of unaffected female progeny; and combined symbols containing numbers the number of unaffected progeny of both sexes. The types of primary cancer and the age (in years) at diagnosis are listed below the symbols, and the bottom-most number is the current age or the age at death. Inf denotes in infancy. Lynch, H. T. et al. N Engl J Med 2003;348:")
204
Initial (Panel A) and Subsequent (Panels B, C, and D) Evaluations of a Pedigree with Classic Hereditary Nonpolyposis Colorectal Cancer Figure 4. Initial (Panel A) and Subsequent (Panels B, C, and D) Evaluations of a Pedigree with Classic Hereditary Nonpolyposis Colorectal Cancer. Panel A shows the initial assessment of what turned out to be a family with classic hereditary nonpolyposis colorectal cancer (HNPCC). The proband (Subject IV-1; arrow) had early-onset (40 years) colorectal carcinoma and carcinoma of the ureter. These findings by themselves are highly significant clinically. However, his mother (Subject III-1) had uterine cervical carcinoma at the age of 37 years, a tumor not associated with the syndrome, but had colorectal carcinoma at the age of 55 years. In Panel B, further inquiry indicated that the proband's mother (Subject III-1) had two sisters with endometrial carcinoma at the ages of 57 (Subject III-2) and 61 (Subject III-3). This pattern, notwithstanding the Amsterdam criteria, would be sufficient for a diagnosis of hereditary nonpolyposis colorectal cancer. In Panel C, extending the pedigree further showed that one of these maternal aunts (Subject III-2) had three sons with cancer, one with early-onset metachronous colon cancer (Subject IV-3), a second with colon cancer and carcinoma of the ureter (Subject IV-4), and the third with colon cancer alone (Subject IV-5). The other aunt (Subject III-3) had a daughter (Subject IV-6) with cancer of the bile duct. These findings provide strong evidence in support of the diagnosis of hereditary nonpolyposis colorectal cancer. In Panel D, the full pedigree shows findings that continue to support a diagnosis of hereditary nonpolyposis colorectal cancer.55 Squares denote male family members; circles female family members; symbols with a slash deceased family members, with the age at death (d) given below each symbol; open symbols unaffected family members; bicolored symbols family members with multiple primary cancers; squares containing numbers the number of unaffected male progeny; circles containing numbers the number of unaffected female progeny; and combined symbols containing numbers the number of unaffected progeny of both sexes. The types of primary cancer and the age (in years) at diagnosis are listed below the symbols, and the bottom-most number is the current age or the age at death. Inf denotes in infancy. Lynch, H. T. et al. N Engl J Med 2003;348:
 and Subsequent (Panels B, C, and D) Evaluations of a Pedigree with Classic Hereditary Nonpolyposis Colorectal Cancer. Panel A shows the initial assessment of what turned out to be a family with classic hereditary nonpolyposis colorectal cancer (HNPCC). The proband (Subject IV-1; arrow) had early-onset (40 years) colorectal carcinoma and carcinoma of the ureter. These findings by themselves are highly significant clinically. However, his mother (Subject III-1) had uterine cervical carcinoma at the age of 37 years, a tumor not associated with the syndrome, but had colorectal carcinoma at the age of 55 years. In Panel B, further inquiry indicated that the proband s mother (Subject III-1) had two sisters with endometrial carcinoma at the ages of 57 (Subject III-2) and 61 (Subject III-3). This pattern, notwithstanding the Amsterdam criteria, would be sufficient for a diagnosis of hereditary nonpolyposis colorectal cancer. In Panel C, extending the pedigree further showed that one of these maternal aunts (Subject III-2) had three sons with cancer, one with early-onset metachronous colon cancer (Subject IV-3), a second with colon cancer and carcinoma of the ureter (Subject IV-4), and the third with colon cancer alone (Subject IV-5). The other aunt (Subject III-3) had a daughter (Subject IV-6) with cancer of the bile duct. These findings provide strong evidence in support of the diagnosis of hereditary nonpolyposis colorectal cancer. In Panel D, the full pedigree shows findings that continue to support a diagnosis of hereditary nonpolyposis colorectal cancer.55. Squares denote male family members; circles female family members; symbols with a slash deceased family members, with the age at death (d) given below each symbol; open symbols unaffected family members; bicolored symbols family members with multiple primary cancers; squares containing numbers the number of unaffected male progeny; circles containing numbers the number of unaffected female progeny; and combined symbols containing numbers the number of unaffected progeny of both sexes. The types of primary cancer and the age (in years) at diagnosis are listed below the symbols, and the bottom-most number is the current age or the age at death. Inf denotes in infancy. Lynch, H. T. et al. N Engl J Med 2003;348:")
205
The Genetics of Breast Cancer
Figure 1. The Genetics of Breast Cancer. BRCA1 and BRCA2 mutations occur in approximately 20 percent of families with evidence of inherited susceptibility to breast cancer. Germ-line mutations in TP53 cause the Li-Fraumeni syndrome and account for no more than 1 percent of cases of familial breast cancer, but women who survive the childhood cancers associated with the Li-Fraumeni syndrome have as much as a 90 percent risk of breast cancer.8 Mutations in the cell-cycle-checkpoint kinase gene (CHEK2) account for about 5 percent of all cases of familial breast cancer (defined by the diagnosis of breast cancer in two or more family members before the age of 60 years), but the risk for individual mutation carriers is probably less than 20 percent.9 All other cases of breast cancer are presumed to be due to an undefined number of additional susceptibility genes with various degrees of penetrance, exposure to hormonal and environmental factors, and stochastic genetic events. Wooster, R. et al. N Engl J Med 2003;348:
 account for about 5 percent of all cases of familial breast cancer (defined by the diagnosis of breast cancer in two or more family members before the age of 60 years), but the risk for individual mutation carriers is probably less than 20 percent.9 All other cases of breast cancer are presumed to be due to an undefined number of additional susceptibility genes with various degrees of penetrance, exposure to hormonal and environmental factors, and stochastic genetic events. Wooster, R. et al. N Engl J Med 2003;348:")
206
CÁNCER: pérdida de control del ciclo celular geneticamente determinada
NO ES UNA ENFERMEDAD HEREDITARIA PUEDE EXISTIR PREDISPOSICIÓN CONSTITUCIONAL PUEDE CONSIDERARSE UNA ENFERMEDAD GENÉTICA

207
EL CÁNCER PUEDE CONSIDERARSE UNA ENFERMEDAD GENÉTICA
Puede resultar de VARIAS MUTACIONES EN CÉLULAS SOMÁTICAS o de la COMBINACIÓN de una MUTACIÓN CONSTITUCIONAL que confiere suceptibilidad a la que se añade una MUTACIÓN SOMÁTICA

208
EL CÁNCER PUEDE CONSIDERARSE UNA ENFERMEDAD GENÉTICA
Puede resultar de VARIAS MUTACIONES EN CÉLULAS SOMÁTICAS o de la COMBINACIÓN de una MUTACIÓN CONSTITUCIONAL que confiere suceptibilidad a la que se añade una MUTACIÓN SOMÁTICA

209
CÁNCER: pérdida de control del ciclo celular geneticamente determinada
VARIAS MUTACIONES QUE AFECTEN A TELOMERASA, FACTORES DE CRECIMIENTO O FACTORES DE TRANSCRIPCIÓN PUEDEN ORIGINAR CÁNCER.

210
Colon Carcinogenesis and the Effects of Chemopreventive Agents
Figure 1. Colon Carcinogenesis and the Effects of Chemopreventive Agents. Colon cancers result from a series of pathologic changes that transform normal colonic epithelium into invasive carcinoma. Specific genetic events, shown by vertical arrows, accompany this multistep process.8 The various chemopreventive agents exert their effects at different steps in this pathway, and this is depicted on the basis of the available epidemiologic evidence, the results of studies in animals, and the known mechanisms of action of the agents. NSAIDs denotes nonsteroidal antiinflammatory drugs, COX-2 cyclooxygenase-2, and APC the adenomatous polyposis coli gene. Janne, P. A. et al. N Engl J Med 2000;342:

211
Colon Carcinogenesis and the Effects of Chemopreventive Agents
Figure 1. Colon Carcinogenesis and the Effects of Chemopreventive Agents. Colon cancers result from a series of pathologic changes that transform normal colonic epithelium into invasive carcinoma. Specific genetic events, shown by vertical arrows, accompany this multistep process.8 The various chemopreventive agents exert their effects at different steps in this pathway, and this is depicted on the basis of the available epidemiologic evidence, the results of studies in animals, and the known mechanisms of action of the agents. NSAIDs denotes nonsteroidal antiinflammatory drugs, COX-2 cyclooxygenase-2, and APC the adenomatous polyposis coli gene. Janne, P. A. et al. N Engl J Med 2000;342:

212
Colon Carcinogenesis and the Effects of Chemopreventive Agents
Figure 1. Colon Carcinogenesis and the Effects of Chemopreventive Agents. Colon cancers result from a series of pathologic changes that transform normal colonic epithelium into invasive carcinoma. Specific genetic events, shown by vertical arrows, accompany this multistep process.8 The various chemopreventive agents exert their effects at different steps in this pathway, and this is depicted on the basis of the available epidemiologic evidence, the results of studies in animals, and the known mechanisms of action of the agents. NSAIDs denotes nonsteroidal antiinflammatory drugs, COX-2 cyclooxygenase-2, and APC the adenomatous polyposis coli gene. Janne, P. A. et al. N Engl J Med 2000;342:

213
Colon Carcinogenesis and the Effects of Chemopreventive Agents
Figure 1. Colon Carcinogenesis and the Effects of Chemopreventive Agents. Colon cancers result from a series of pathologic changes that transform normal colonic epithelium into invasive carcinoma. Specific genetic events, shown by vertical arrows, accompany this multistep process.8 The various chemopreventive agents exert their effects at different steps in this pathway, and this is depicted on the basis of the available epidemiologic evidence, the results of studies in animals, and the known mechanisms of action of the agents. NSAIDs denotes nonsteroidal antiinflammatory drugs, COX-2 cyclooxygenase-2, and APC the adenomatous polyposis coli gene. Janne, P. A. et al. N Engl J Med 2000;342:

214
EL CÁNCER PUEDE CONSIDERARSE UNA ENFERMEDAD GENÉTICA
Puede resultar de VARIAS MUTACIONES EN CÉLULAS SOMÁTICAS o de la COMBINACIÓN de una MUTACIÓN CONSTITUCIONAL que confiere suceptibilidad a la que se añade una MUTACIÓN SOMÁTICA

215
BASES GENÉTICAS DEL CANCER. II
Patología General 21 de Septiembre de 2015 Remigio Cordero Torres

216
EL CÁNCER PUEDE CONSIDERARSE UNA ENFERMEDAD GENÉTICA
Puede resultar de VARIAS MUTACIONES EN CÉLULAS SOMÁTICAS o de la COMBINACIÓN de una MUTACIÓN CONSTITUCIONAL que confiere suceptibilidad a la que se añade una MUTACIÓN SOMÁTICA

217
GENES RELACIONADOS CON CÁNCER
ONCOGENES GENES SUPRESORES DE TUMORES GENES DE REPARACIÓN DE ERRORES microRNAs

218
GENES RELACIONADOS CON CÁNCER
ONCOGENES PROTOONCOGENES: genes que controlan división celular. Se activan cuando son necesarias altas tasas de división celular ONCOGEN: protooncogen activado en tiempo o lugar equivocado Codifican las ONCOPROTEÍNAS Se comportan de forma DOMINANTE: es suficiente con la lesión de uno de los dos alelos del correspondiente gen para que la producción excesiva de la oncoproteína acelere de forma desordenada el crecimiento celular

219
Cyclin D1 and the Development of Breast Cancer
Figure 1. Cyclin D1 and the Development of Breast Cancer. Cyclin D1 has a unique role in regulating the cell cycle in alveolar epithelial cells of the breast (Panel A). It is a possible therapeutic target because it is overexpressed in breast cancer, especially estrogen-receptor-positive breast cancers. Trastuzumab, the clinically useful monoclonal antibody against NEU, may indirectly inhibit cyclin D1 through the RAS pathway. In mice, the actions of the Neu and Ras oncogenes in the breast depend on cyclin D1, as suggested in Panel B. Mammary cancer induced in mice by overexpression of the Neu and Ras oncogenes depends on cyclin D1, whereas the Myc and Wnt1 oncogenes can act independently of cyclin D1. Chodosh, L. A. N Engl J Med 2002;347:
. It is a possible therapeutic target because it is overexpressed in breast cancer, especially estrogen-receptor-positive breast cancers. Trastuzumab, the clinically useful monoclonal antibody against NEU, may indirectly inhibit cyclin D1 through the RAS pathway. In mice, the actions of the Neu and Ras oncogenes in the breast depend on cyclin D1, as suggested in Panel B. Mammary cancer induced in mice by overexpression of the Neu and Ras oncogenes depends on cyclin D1, whereas the Myc and Wnt1 oncogenes can act independently of cyclin D1. Chodosh, L. A. N Engl J Med 2002;347:")
220
ONCOGENES FACTORES DE CRECIMIENTO
Oncogen sys: factor crecimiento derivado de plaquetas RECEPTORES DE FACTORES DE CRECIMIENTO Oncogen erb: receptores factor crecimiento epidérmico Oncogen ret: receptor acividad tirosin cinasa VÍAS TRANSDUCCIÓN DE SEÑALES AL INTERIOR CELULAR Oncogen ras Oncogen abl FACTORES DE TRANSCRIPCIÓN Protooncogenes myc, jun, fos, myb. PROTEINAS REGULADORAS DEL CICLO CELULAR Ciclinas D o CD4

221
Mecanismos de activación de oncogenes
AUMENTO EXPRESIÓN NUEVA LOCALIZACIÓN. VIRUS INVERSIÓN TRANSLOCACIÓN VEB FUSIÓN DE PROTEÍNAS CON NUEVAS FUNCIONES LMC 9->22 ABL Y BCR L PROMIELOCÍTICA AGUDA 15->17. Receptor ácido retinoico – oncogen myc

222
A Potential Mechanism of Leukemogenesis
Figure 1. A Potential Mechanism of Leukemogenesis. T-cell leukemia developed in two children with SCID who were treated with CD34 stem cells transduced with a viral vector containing IL2RG, which encodes the common {gamma} subunit of the interleukin-2 receptor. A recent study by Dave et al.3 suggests that the{gamma} c transgene was inserted close to the LMO2 gene, activating the T-cell oncogene. Overexpression of the LMO2 protein in T cells blocks the differentiation of the cells and thus increases their susceptibility to leukemia. This finding has potential implications for the design of gene-therapy protocols. A Potential Mechanism of Leukemogenesis Berns, A. N Engl J Med 2004;350:

223
Figure 3. Translocation Leading to the Philadelphia (Ph) Chromosome and the Role of BCR-ABL in the Pathogenesis of CML (Panel A) and the Effect of Normal (Panel B) and Abnormal (Panel C) c-kit Function on Platelet-Derived Growth Factor and Gastrointestinal Stromal Tumors. The Ph chromosome is a foreshortened chromosome 22 resulting from an exchange between the long arms of chromosomes 9 and 22 (Panel A). The translocation -- t(9;22) -- results in the juxtaposition of 3' DNA sequences derived from the ABL proto-oncogene on chromosome 9 with 5' sequences of the breakpoint cluster region (BCR) gene on chromosome 22, forming a fusion gene, BCR-ABL. (The reciprocal formation of the ABL-BCR fusion gene on chromosome 9q is not depicted.) BCR-ABL produces a chimeric messenger RNA (not shown) from which a fusion BCR-ABL oncoprotein is translated. The length of the BCR-ABL protein varies and is determined by the breakpoint within the BCR gene. Chronic-phase CML is driven by the constitutively active BCR-ABL tyrosine kinase protein, which activates multiple pathways, leading to the malignant expansion of myeloid cells through the stimulation of mitosis, the disruption of cytoadherence and regulatory control by stromal cells, and the inhibition of apoptosis. Differentiation and maturation of the leukemic clone are relatively intact in chronic-phase CML, but BCR-ABL is also thought to promote genomic instability, leading to secondary mutations and to the blast phase. Imatinib mesylate inhibits the tyrosine kinase activity of the BCR-ABL oncoprotein, thus blocking the leukemogenic effects of the Ph chromosome. Dimerization and activation of the normal c-kit receptor by its ligand stem-cell factor are shown in Panel B. The proto-oncogene c-kit encodes a transmembrane tyrosine kinase receptor located on the long arm of chromosome 4 (4q11-q12). In gastrointestinal stromal tumors, in-frame deletions and point mutations in c-kit produce ligand-independent constitutive activation of c-kit (Panel C). Mutations of c-kit in the juxtamembrane domain in gastrointestinal stromal tumors (exon 11) are found in approximately 60 percent of cases. Mutations also occur in the extracellular domain (exon 9) and in the more distal phosphokinase domain (exon 13). ATP denotes adenosine triphosphate, ADP adenosine diphosphate, and P phosphate. Savage, D.NEJM 2002 ;346: Translocation Leading to the Philadelphia (Ph) Chromosome and the Role of BCR-ABL in the Pathogenesis of CML
. The translocation -- t(9;22) -- results in the juxtaposition of 3 DNA sequences derived from the ABL proto-oncogene on chromosome 9 with 5 sequences of the breakpoint cluster region (BCR) gene on chromosome 22, forming a fusion gene, BCR-ABL. (The reciprocal formation of the ABL-BCR fusion gene on chromosome 9q is not depicted.) BCR-ABL produces a chimeric messenger RNA (not shown) from which a fusion BCR-ABL oncoprotein is translated. The length of the BCR-ABL protein varies and is determined by the breakpoint within the BCR gene. Chronic-phase CML is driven by the constitutively active BCR-ABL tyrosine kinase protein, which activates multiple pathways, leading to the malignant expansion of myeloid cells through the stimulation of mitosis, the disruption of cytoadherence and regulatory control by stromal cells, and the inhibition of apoptosis. Differentiation and maturation of the leukemic clone are relatively intact in chronic-phase CML, but BCR-ABL is also thought to promote genomic instability, leading to secondary mutations and to the blast phase. Imatinib mesylate inhibits the tyrosine kinase activity of the BCR-ABL oncoprotein, thus blocking the leukemogenic effects of the Ph chromosome. Dimerization and activation of the normal c-kit receptor by its ligand stem-cell factor are shown in Panel B. The proto-oncogene c-kit encodes a transmembrane tyrosine kinase receptor located on the long arm of chromosome 4 (4q11-q12). In gastrointestinal stromal tumors, in-frame deletions and point mutations in c-kit produce ligand-independent constitutive activation of c-kit (Panel C). Mutations of c-kit in the juxtamembrane domain in gastrointestinal stromal tumors (exon 11) are found in approximately 60 percent of cases. Mutations also occur in the extracellular domain (exon 9) and in the more distal phosphokinase domain (exon 13). ATP denotes adenosine triphosphate, ADP adenosine diphosphate, and P phosphate. Savage, D.NEJM 2002 ;346: Translocation Leading to the Philadelphia (Ph) Chromosome and the Role of BCR-ABL in the Pathogenesis of CML.")
224
Translocation Leading to the Philadelphia (Ph) Chromosome and the Role of BCR-ABL in the Pathogenesis of CML (Panel A) and the Effect of Normal (Panel B) and Abnormal (Panel C) c-kit Function on Platelet-Derived Growth Factor and Gastrointestinal Stromal Tumors Figure 3. Translocation Leading to the Philadelphia (Ph) Chromosome and the Role of BCR-ABL in the Pathogenesis of CML (Panel A) and the Effect of Normal (Panel B) and Abnormal (Panel C) c-kit Function on Platelet-Derived Growth Factor and Gastrointestinal Stromal Tumors. The Ph chromosome is a foreshortened chromosome 22 resulting from an exchange between the long arms of chromosomes 9 and 22 (Panel A). The translocation -- t(9;22) -- results in the juxtaposition of 3' DNA sequences derived from the ABL proto-oncogene on chromosome 9 with 5' sequences of the breakpoint cluster region (BCR) gene on chromosome 22, forming a fusion gene, BCR-ABL. (The reciprocal formation of the ABL-BCR fusion gene on chromosome 9q is not depicted.) BCR-ABL produces a chimeric messenger RNA (not shown) from which a fusion BCR-ABL oncoprotein is translated. The length of the BCR-ABL protein varies and is determined by the breakpoint within the BCR gene. Chronic-phase CML is driven by the constitutively active BCR-ABL tyrosine kinase protein, which activates multiple pathways, leading to the malignant expansion of myeloid cells through the stimulation of mitosis, the disruption of cytoadherence and regulatory control by stromal cells, and the inhibition of apoptosis. Differentiation and maturation of the leukemic clone are relatively intact in chronic-phase CML, but BCR-ABL is also thought to promote genomic instability, leading to secondary mutations and to the blast phase. Imatinib mesylate inhibits the tyrosine kinase activity of the BCR-ABL oncoprotein, thus blocking the leukemogenic effects of the Ph chromosome. Dimerization and activation of the normal c-kit receptor by its ligand stem-cell factor are shown in Panel B. The proto-oncogene c-kit encodes a transmembrane tyrosine kinase receptor located on the long arm of chromosome 4 (4q11-q12). In gastrointestinal stromal tumors, in-frame deletions and point mutations in c-kit produce ligand-independent constitutive activation of c-kit (Panel C). Mutations of c-kit in the juxtamembrane domain in gastrointestinal stromal tumors (exon 11) are found in approximately 60 percent of cases. Mutations also occur in the extracellular domain (exon 9) and in the more distal phosphokinase domain (exon 13). ATP denotes adenosine triphosphate, ADP adenosine diphosphate, and P phosphate. Savage, D. G. et al. N Engl J Med 2002;346:
 Chromosome and the Role of BCR-ABL in the Pathogenesis of CML (Panel A) and the Effect of Normal (Panel B) and Abnormal (Panel C) c-kit Function on Platelet-Derived Growth Factor and Gastrointestinal Stromal Tumors. The Ph chromosome is a foreshortened chromosome 22 resulting from an exchange between the long arms of chromosomes 9 and 22 (Panel A). The translocation -- t(9;22) -- results in the juxtaposition of 3 DNA sequences derived from the ABL proto-oncogene on chromosome 9 with 5 sequences of the breakpoint cluster region (BCR) gene on chromosome 22, forming a fusion gene, BCR-ABL. (The reciprocal formation of the ABL-BCR fusion gene on chromosome 9q is not depicted.) BCR-ABL produces a chimeric messenger RNA (not shown) from which a fusion BCR-ABL oncoprotein is translated. The length of the BCR-ABL protein varies and is determined by the breakpoint within the BCR gene. Chronic-phase CML is driven by the constitutively active BCR-ABL tyrosine kinase protein, which activates multiple pathways, leading to the malignant expansion of myeloid cells through the stimulation of mitosis, the disruption of cytoadherence and regulatory control by stromal cells, and the inhibition of apoptosis. Differentiation and maturation of the leukemic clone are relatively intact in chronic-phase CML, but BCR-ABL is also thought to promote genomic instability, leading to secondary mutations and to the blast phase. Imatinib mesylate inhibits the tyrosine kinase activity of the BCR-ABL oncoprotein, thus blocking the leukemogenic effects of the Ph chromosome. Dimerization and activation of the normal c-kit receptor by its ligand stem-cell factor are shown in Panel B. The proto-oncogene c-kit encodes a transmembrane tyrosine kinase receptor located on the long arm of chromosome 4 (4q11-q12). In gastrointestinal stromal tumors, in-frame deletions and point mutations in c-kit produce ligand-independent constitutive activation of c-kit (Panel C). Mutations of c-kit in the juxtamembrane domain in gastrointestinal stromal tumors (exon 11) are found in approximately 60 percent of cases. Mutations also occur in the extracellular domain (exon 9) and in the more distal phosphokinase domain (exon 13). ATP denotes adenosine triphosphate, ADP adenosine diphosphate, and P phosphate. Savage, D. G. et al. N Engl J Med 2002;346:")
225
Translocation Leading to the Philadelphia (Ph) Chromosome and the Role of BCR-ABL in the Pathogenesis of CML (Panel A) and the Effect of Normal (Panel B) and Abnormal (Panel C) c-kit Function on Platelet-Derived Growth Factor and Gastrointestinal Stromal Tumors Figure 3. Translocation Leading to the Philadelphia (Ph) Chromosome and the Role of BCR-ABL in the Pathogenesis of CML (Panel A) and the Effect of Normal (Panel B) and Abnormal (Panel C) c-kit Function on Platelet-Derived Growth Factor and Gastrointestinal Stromal Tumors. The Ph chromosome is a foreshortened chromosome 22 resulting from an exchange between the long arms of chromosomes 9 and 22 (Panel A). The translocation -- t(9;22) -- results in the juxtaposition of 3' DNA sequences derived from the ABL proto-oncogene on chromosome 9 with 5' sequences of the breakpoint cluster region (BCR) gene on chromosome 22, forming a fusion gene, BCR-ABL. (The reciprocal formation of the ABL-BCR fusion gene on chromosome 9q is not depicted.) BCR-ABL produces a chimeric messenger RNA (not shown) from which a fusion BCR-ABL oncoprotein is translated. The length of the BCR-ABL protein varies and is determined by the breakpoint within the BCR gene. Chronic-phase CML is driven by the constitutively active BCR-ABL tyrosine kinase protein, which activates multiple pathways, leading to the malignant expansion of myeloid cells through the stimulation of mitosis, the disruption of cytoadherence and regulatory control by stromal cells, and the inhibition of apoptosis. Differentiation and maturation of the leukemic clone are relatively intact in chronic-phase CML, but BCR-ABL is also thought to promote genomic instability, leading to secondary mutations and to the blast phase. Imatinib mesylate inhibits the tyrosine kinase activity of the BCR-ABL oncoprotein, thus blocking the leukemogenic effects of the Ph chromosome. Dimerization and activation of the normal c-kit receptor by its ligand stem-cell factor are shown in Panel B. The proto-oncogene c-kit encodes a transmembrane tyrosine kinase receptor located on the long arm of chromosome 4 (4q11-q12). In gastrointestinal stromal tumors, in-frame deletions and point mutations in c-kit produce ligand-independent constitutive activation of c-kit (Panel C). Mutations of c-kit in the juxtamembrane domain in gastrointestinal stromal tumors (exon 11) are found in approximately 60 percent of cases. Mutations also occur in the extracellular domain (exon 9) and in the more distal phosphokinase domain (exon 13). ATP denotes adenosine triphosphate, ADP adenosine diphosphate, and P phosphate. Savage, D. G. et al. N Engl J Med 2002;346:
 Chromosome and the Role of BCR-ABL in the Pathogenesis of CML (Panel A) and the Effect of Normal (Panel B) and Abnormal (Panel C) c-kit Function on Platelet-Derived Growth Factor and Gastrointestinal Stromal Tumors. The Ph chromosome is a foreshortened chromosome 22 resulting from an exchange between the long arms of chromosomes 9 and 22 (Panel A). The translocation -- t(9;22) -- results in the juxtaposition of 3 DNA sequences derived from the ABL proto-oncogene on chromosome 9 with 5 sequences of the breakpoint cluster region (BCR) gene on chromosome 22, forming a fusion gene, BCR-ABL. (The reciprocal formation of the ABL-BCR fusion gene on chromosome 9q is not depicted.) BCR-ABL produces a chimeric messenger RNA (not shown) from which a fusion BCR-ABL oncoprotein is translated. The length of the BCR-ABL protein varies and is determined by the breakpoint within the BCR gene. Chronic-phase CML is driven by the constitutively active BCR-ABL tyrosine kinase protein, which activates multiple pathways, leading to the malignant expansion of myeloid cells through the stimulation of mitosis, the disruption of cytoadherence and regulatory control by stromal cells, and the inhibition of apoptosis. Differentiation and maturation of the leukemic clone are relatively intact in chronic-phase CML, but BCR-ABL is also thought to promote genomic instability, leading to secondary mutations and to the blast phase. Imatinib mesylate inhibits the tyrosine kinase activity of the BCR-ABL oncoprotein, thus blocking the leukemogenic effects of the Ph chromosome. Dimerization and activation of the normal c-kit receptor by its ligand stem-cell factor are shown in Panel B. The proto-oncogene c-kit encodes a transmembrane tyrosine kinase receptor located on the long arm of chromosome 4 (4q11-q12). In gastrointestinal stromal tumors, in-frame deletions and point mutations in c-kit produce ligand-independent constitutive activation of c-kit (Panel C). Mutations of c-kit in the juxtamembrane domain in gastrointestinal stromal tumors (exon 11) are found in approximately 60 percent of cases. Mutations also occur in the extracellular domain (exon 9) and in the more distal phosphokinase domain (exon 13). ATP denotes adenosine triphosphate, ADP adenosine diphosphate, and P phosphate. Savage, D. G. et al. N Engl J Med 2002;346:")
226
GENES RELACIONADOS CON CÁNCER
ONCOGENES GENES SUPRESORES DE TUMORES GENES DE REPARACIÓN DE ERRORES microRNAs

227
GENES RELACIONADOS CON CÁNCER
ONCOGENES GENES SUPRESORES DE TUMORES GENES DE REPARACIÓN DE ERRORES microRNAs

228
GENES SUPRESORES DE TUMORES
Su función fisiológica es controlar el crecimiento celular. Su inactivación y pérdida de función desencadena el proceso neoplásico Se requiere la afectación de los dos alelos del gen (Comportamiento recesivo en la carcinogénesis)
")
229
GENES SUPRESORES DE TUMORES
RETINOBLASTOMA 1 mutación heredada + 1 mutación somática en 13q Función normal pRB : secuestrar factor de transcripción E2F Pérdida función-> E2F libre-> activación transcripción GEN P53 WT1 (Wilms) NF1 Y NF2 (Neurofibromatosis) DCC (Cáncer de colon) APC (Poliposis adenomatosa familiar)
 NF1 Y NF2 (Neurofibromatosis) DCC (Cáncer de colon) APC (Poliposis adenomatosa familiar)")
230
Genetic and Epigenetic Changes That Inactivate Tumor-Suppressor Genes According to the Knudson Two-Hit Hypothesis Figure 3. Genetic and Epigenetic Changes That Inactivate Tumor-Suppressor Genes According to the Knudson Two-Hit Hypothesis. The first hit can be a coding-region mutation in a tumor-suppressor gene, shown on the short arm of the green chromosome. This can occur as either a germ-line event, in inherited cancers, or a somatic event, in sporadic cancers. Alternatively, promoter hypermethylation and associated silencing of gene transcription can constitute the first hit in somatic cancers but not in inherited cancers. Often, in somatic cancers, chromosomal deletions (absent regions in the short arm of the depicted chromosomes) involving the second gene copy constitute the second hit to eliminate the remaining gene copy. It is rare in such tumors to have point mutations constitute the hits in both copies of a tumor-suppressor gene, but hypermethylation of both copies is not infrequently found. In inherited cancers, with germ-line mutations in one copy of the tumor-suppressor gene, the second hit in the tumors can consist of either chromosomal deletions or promoter hypermethylation. Herman, J. G. et al. N Engl J Med 2003;349:
 involving the second gene copy constitute the second hit to eliminate the remaining gene copy. It is rare in such tumors to have point mutations constitute the hits in both copies of a tumor-suppressor gene, but hypermethylation of both copies is not infrequently found. In inherited cancers, with germ-line mutations in one copy of the tumor-suppressor gene, the second hit in the tumors can consist of either chromosomal deletions or promoter hypermethylation. Herman, J. G. et al. N Engl J Med 2003;349:")
231
Cambios epigenéticos Cambios heredables en la estructura de un gen sin alterar la secuencia del DNA. Los dos más frecuentes son la METILACIÓN y la modificación de las histonas por acetilación. METILACIÓN: Unión de grupo metilo al carbono 5 en una isla CpG (región de 500 pares de bases con alto contenido en Citosina y Guanina). Hipermetilación : silenciamiento de genes. Hipometilación: nivel de transcripción elevado
. Hipermetilación : silenciamiento de genes. Hipometilación: nivel de transcripción elevado.")
232
Epigenetic Alterations in Tumor Progression
Figure 1. Epigenetic Alterations in Tumor Progression. A multistage model of carcinogenesis in skin is shown. In conjunction with phenotypic cellular changes and the accumulation of genetic defects, there is a progressive loss of total DNA methylation content, an increased frequency of hypermethylated CpG islands, and an increased histone-modification imbalance in the development of the disease. H-ras denotes Harvey-ras oncogene, and 5mC 5-methylcytosine. Esteller M. N Engl J Med 2008;358:

233
GENES SUPRESORES DE TUMORES
RETINOBLASTOMA GEN P53 Delección en 17p: pérdida de producción de proteína p53 Prot P53: detecta daño ADN. Interrumpe ciclo en G1. Repara o provoca muerte celular (apoptosis) si daño grave En la mayoría de los casos mutaciones somáticas. Mutación en linea germinal (LI FRAUMENI) 100 familias WT1 (Wilms) NF1 Y NF2 (Neurofibromatosis) DCC (Cáncer de colon) APC (Poliposis adenomatosa familiar)
 si daño grave. En la mayoría de los casos mutaciones somáticas. Mutación en linea germinal (LI FRAUMENI) 100 familias. WT1 (Wilms) NF1 Y NF2 (Neurofibromatosis) DCC (Cáncer de colon) APC (Poliposis adenomatosa familiar)")
234
Mutations in Genes That Regulate Cellular Proliferation and the Repair of DNA and Lead to the Development of Breast Cancer Haber, D. N Engl J Med 2000;343: Figure 1. Mutations in Genes That Regulate Cellular Proliferation and the Repair of DNA and Lead to the Development of Breast Cancer. The ATM (ataxia-telangiectasia mutated) gene encodes a kinase that phosphorylates and thus activates the tumor-suppressor p53 either directly or by activating CHK2 in response to damage to DNA. The p53 protein then triggers the arrest of the cell cycle, allowing time for DNA to be repaired. The BRCA1 and NBS proteins are also activated by ATM and are thought to be directly involved in the repair of damaged DNA. The inactivating mutations in the genes that encode these proteins and thus lead to or increase the risk of breast cancer are shown. G1 and G2 are gap 1 and 2.
 gene encodes a kinase that phosphorylates and thus activates the tumor-suppressor p53 either directly or by activating CHK2 in response to damage to DNA. The p53 protein then triggers the arrest of the cell cycle, allowing time for DNA to be repaired. The BRCA1 and NBS proteins are also activated by ATM and are thought to be directly involved in the repair of damaged DNA. The inactivating mutations in the genes that encode these proteins and thus lead to or increase the risk of breast cancer are shown. G1 and G2 are gap 1 and 2.")
235
GENES RELACIONADOS CON CÁNCER
ONCOGENES GENES SUPRESORES DE TUMORES GENES DE REPARACIÓN DE ERRORES microRNAs

236
Sources and Consequences of DNA Damage
Figure 1. Sources and Consequences of DNA Damage. DNA damage can be induced by exogenous physical agents, by endogenous chemical genotoxic agents that are the products of metabolism, such as reactive oxygen species (ROS), or by spontaneous chemical reactions, such as hydrolysis. Examples of DNA damage are ultraviolet (UV)-induced photoproducts (left), interstrand and intrastrand crosslinks, bulky chemical adducts (purple sphere), abasic sites, and oxidative damage such as 8-oxoguanine (8-oxoG). The consequences of DNA damage are essentially twofold. After misrepair or replication of the damaged template, surviving cells may be subject to permanent changes in the genetic code in the form of mutations or chromosomal aberrations, both of which increase the risk of cancer. Alternatively, damage may interfere with the vital process of transcription or induce replication arrest, which may trigger cell death or cellular senescence, contributing to aging. Damage-induced cell death protects the body from cancer. G denotes guanine, and T thymidine. Hoeijmakers J. N Engl J Med 2009;361:
, or by spontaneous chemical reactions, such as hydrolysis. Examples of DNA damage are ultraviolet (UV)-induced photoproducts (left), interstrand and intrastrand crosslinks, bulky chemical adducts (purple sphere), abasic sites, and oxidative damage such as 8-oxoguanine (8-oxoG). The consequences of DNA damage are essentially twofold. After misrepair or replication of the damaged template, surviving cells may be subject to permanent changes in the genetic code in the form of mutations or chromosomal aberrations, both of which increase the risk of cancer. Alternatively, damage may interfere with the vital process of transcription or induce replication arrest, which may trigger cell death or cellular senescence, contributing to aging. Damage-induced cell death protects the body from cancer. G denotes guanine, and T thymidine. Hoeijmakers J. N Engl J Med 2009;361:")
237
Mutations in Genes That Regulate Cellular Proliferation and the Repair of DNA and Lead to the Development of Breast Cancer Haber, D. N Engl J Med 2000;343: Figure 1. Mutations in Genes That Regulate Cellular Proliferation and the Repair of DNA and Lead to the Development of Breast Cancer. The ATM (ataxia-telangiectasia mutated) gene encodes a kinase that phosphorylates and thus activates the tumor-suppressor p53 either directly or by activating CHK2 in response to damage to DNA. The p53 protein then triggers the arrest of the cell cycle, allowing time for DNA to be repaired. The BRCA1 and NBS proteins are also activated by ATM and are thought to be directly involved in the repair of damaged DNA. The inactivating mutations in the genes that encode these proteins and thus lead to or increase the risk of breast cancer are shown. G1 and G2 are gap 1 and 2.
 gene encodes a kinase that phosphorylates and thus activates the tumor-suppressor p53 either directly or by activating CHK2 in response to damage to DNA. The p53 protein then triggers the arrest of the cell cycle, allowing time for DNA to be repaired. The BRCA1 and NBS proteins are also activated by ATM and are thought to be directly involved in the repair of damaged DNA. The inactivating mutations in the genes that encode these proteins and thus lead to or increase the risk of breast cancer are shown. G1 and G2 are gap 1 and 2.")
238
Cancer-Susceptibility Genes and DNA Repair
Figure 1. Cancer-Susceptibility Genes and DNA Repair. Several genes (ATM, CHEK2, BRCA1, and BRCA2) whose inactivation predisposes people to breast and other cancers participate in the error-free repair of breaks in double-stranded DNA by homologous recombination. The process starts when ATM and CHEK2 protein kinases signal the presence of double-stranded breaks by phosphorylating (red arrows) proteins such as BRCA1, inducing their migration to sites where DNA is repaired. BRCA2 carries the DNA-recombination enzyme RAD51 to the same sites. It is guided there by the DNA-binding structures formed between its carboxy terminal and Dss1. The concerted activity of these proteins culminates in error-free DNA repair by recombination. Two findings1,2 connect Fanconi's anemia proteins to this pathway. First, a complex of Fanconi's anemia proteins -- termed A, C, D2, E, F, and G -- triggers the ubiquitination of the D2 protein alone and its colocalization with BRCA1. Second, BRCA2 is mutated in a small group of patients with Fanconi's anemia. This work emphasizes the importance of the homologous recombination pathway in the pathogenesis of disorders involving chromosomal instability. Ub denotes mono-ubiquitin. Venkitaraman, A. R. N Engl J Med 2003;348:
 whose inactivation predisposes people to breast and other cancers participate in the error-free repair of breaks in double-stranded DNA by homologous recombination. The process starts when ATM and CHEK2 protein kinases signal the presence of double-stranded breaks by phosphorylating (red arrows) proteins such as BRCA1, inducing their migration to sites where DNA is repaired. BRCA2 carries the DNA-recombination enzyme RAD51 to the same sites. It is guided there by the DNA-binding structures formed between its carboxy terminal and Dss1. The concerted activity of these proteins culminates in error-free DNA repair by recombination. Two findings1,2 connect Fanconi s anemia proteins to this pathway. First, a complex of Fanconi s anemia proteins -- termed A, C, D2, E, F, and G -- triggers the ubiquitination of the D2 protein alone and its colocalization with BRCA1. Second, BRCA2 is mutated in a small group of patients with Fanconi s anemia. This work emphasizes the importance of the homologous recombination pathway in the pathogenesis of disorders involving chromosomal instability. Ub denotes mono-ubiquitin. Venkitaraman, A. R. N Engl J Med 2003;348:")
239
The Genetics of Breast Cancer
Figure 1. The Genetics of Breast Cancer. BRCA1 and BRCA2 mutations occur in approximately 20 percent of families with evidence of inherited susceptibility to breast cancer. Germ-line mutations in TP53 cause the Li-Fraumeni syndrome and account for no more than 1 percent of cases of familial breast cancer, but women who survive the childhood cancers associated with the Li-Fraumeni syndrome have as much as a 90 percent risk of breast cancer.8 Mutations in the cell-cycle-checkpoint kinase gene (CHEK2) account for about 5 percent of all cases of familial breast cancer (defined by the diagnosis of breast cancer in two or more family members before the age of 60 years), but the risk for individual mutation carriers is probably less than 20 percent.9 All other cases of breast cancer are presumed to be due to an undefined number of additional susceptibility genes with various degrees of penetrance, exposure to hormonal and environmental factors, and stochastic genetic events. Wooster, R. et al. N Engl J Med 2003;348:
 account for about 5 percent of all cases of familial breast cancer (defined by the diagnosis of breast cancer in two or more family members before the age of 60 years), but the risk for individual mutation carriers is probably less than 20 percent.9 All other cases of breast cancer are presumed to be due to an undefined number of additional susceptibility genes with various degrees of penetrance, exposure to hormonal and environmental factors, and stochastic genetic events. Wooster, R. et al. N Engl J Med 2003;348:")
240
Inestabilidad genética por alteraciones de GENES DE REPARACIÓN DE ERRORES
Enfermedad Frecuencia Defecto Ataxia-Telangiectasia 1/40.000 Kinasa que controla ciclo Síndrome de Bloom 100 casos Inactividad de DNA ligasa Anemia de Fanconi 1/22.000 Reparación por escisión Ca colon no polipósico 1/200 Reparación por malapareamiento Xeroderma pigmentosum 1/

241
GENES RELACIONADOS CON CÁNCER
ONCOGENES GENES SUPRESORES DE TUMORES GENES DE REPARACIÓN DE ERRORES microRNAs

242
MicroRNA No codifican proteínas.
Su producto es una fibra simple de RNA de 21 a 23 nucleótidos. Función: regular la expresión de genes. Puede bloquear la traslación de un RNA mensajero o degradarlo. Puede ser supresores si su diana es un oncogen o se pueden comportar como oncogenes si su diana es un gen supresor Pueden activarse (amplificación) o inactivarse (delección, silenciado epigenético o pérdida de expresión de factores de transcripción)
 o inactivarse (delección, silenciado epigenético o pérdida de expresión de factores de transcripción)")
243
The Increasing Complexity of the Central Dogma of Molecular Biology.
Figure 1. The Increasing Complexity of the Central Dogma of Molecular Biology. The flow of genomic information from DNA to RNA to protein remains the basis for understanding genomic function (Panel A). A single gene can yield an extensive array of gene products, depending on the environment in which it is expressed, thereby expanding the repertoire of the 20,000 or so genes in the human genome (Panel B). The initial event of gene expression, transcription, is regulated by means of a complex choreography of events involving the three-dimensional DNA structure, covalent chemical, or epigenetic, modifications of the DNA backbone, and interactions between protein and DNA and between RNA and DNA. Translation is similarly complex and tightly regulated by interactions between messenger RNA (mRNA) and proteins. Processing of single-precursor RNA (preRNA) molecules can yield multiple RNA products, including microRNA (miRNA) and small interfering RNA (siRNA) molecules. Post-translational modification of proteins also contributes greatly to the diversity of the output of the human genome through modifications of individual immature proteins (e.g., folding, cleavage, and chemical modifications), which yield an array of related protein products. Feero WG et al. N Engl J Med 2010;362:
. A single gene can yield an extensive array of gene products, depending on the environment in which it is expressed, thereby expanding the repertoire of the 20,000 or so genes in the human genome (Panel B). The initial event of gene expression, transcription, is regulated by means of a complex choreography of events involving the three-dimensional DNA structure, covalent chemical, or epigenetic, modifications of the DNA backbone, and interactions between protein and DNA and between RNA and DNA. Translation is similarly complex and tightly regulated by interactions between messenger RNA (mRNA) and proteins. Processing of single-precursor RNA (preRNA) molecules can yield multiple RNA products, including microRNA (miRNA) and small interfering RNA (siRNA) molecules. Post-translational modification of proteins also contributes greatly to the diversity of the output of the human genome through modifications of individual immature proteins (e.g., folding, cleavage, and chemical modifications), which yield an array of related protein products. Feero WG et al. N Engl J Med 2010;362:")
244
CARACTERES DE CÉLULAS DEL CÁNCER
PÉRDIDA CONTROL CICLO CLONALIDAD HETEROGENEIDAD MUTABILIDAD GENÉTICA TRANSPLANTABILIDAD DESDIFERENCIACIÓN PÉRDIDA INHIBICIÓN DE CONTACTO ANGIOGÉNESIS INVASIVIDAD TEJIDOS ADYACENTES METÁSTASIS A DISTANCIA

245
Alteraciones celulares en la evolución de un tumor
Independencia de señales de crecimiento. (Insensibilidad señales inhibitorias crecimiento) Escape de muerte celular programada. Potencial Proliferativo Aumentado. Estimulación de la Angiogénesis. Capacidad invadir tejidos vecinos y lejanos
 Escape de muerte celular programada. Potencial Proliferativo Aumentado. Estimulación de la Angiogénesis. Capacidad invadir tejidos vecinos y lejanos.")
246
Benignidad / Malignidad
Tumores Benignos Tumores Malignos Grado de Diferenciación Bien diferenciados Desdiferenciados Anaplásicos Velocidad de crecimiento Lento Escasas Mitosis Rápido Múltiples mitosis Invasión regional No Encapsulados Sí Infiltrantes Metástasis

247
Tasas de división celular
Células Normales Horas entre división Células precursoras de médula ósea 18 Células recubrimiento intestino delgado 39 Células recubrimiento de recto 48 Óvulos fertilizados 36-60 Células de cáncer Horas entre división Cáncer de Estómago 72 Leucemia mieloblástica aguda 80-84 Leucemia mieloide crónica 120 Cáncer de pulmón

248
Progresión del cáncer CRECIMIENTO TUMORAL ANGIOGÉNESIS
Fracción de crecimiento. Tipo de Tumor (Leucemias, tumores agresivos) Momento evolutivo (Mayor en fases precoces) ANGIOGÉNESIS Degradación MB vaso sanguíneo preexistente Migración células endoteliales Proliferación y remodelación Factor de crecimiento endotelial (VEGF) INVASIÓN REGIONAL- METÁSTASIS
 Momento evolutivo (Mayor en fases precoces) ANGIOGÉNESIS. Degradación MB vaso sanguíneo preexistente. Migración células endoteliales. Proliferación y remodelación. Factor de crecimiento endotelial (VEGF) INVASIÓN REGIONAL- METÁSTASIS.")
250
Invasión regional - metástasis
Pérdida de adherencia celular Defecto en cadherina E (desmosomas) Paso a través de matriz intercelular Unión a matriz intercelular Degradantes matriz (metaloproteínas) Migración (factores de motilidad) Diseminación Estirpe Epitelial (carcinomas): predom vía linfática Estirpe mesenquimal(sarcomas): pred vía sanguinea Metástasis Factores quimiotácticos – Moléculas de adhesión
 Paso a través de matriz intercelular. Unión a matriz intercelular. Degradantes matriz (metaloproteínas) Migración (factores de motilidad) Diseminación. Estirpe Epitelial (carcinomas): predom vía linfática. Estirpe mesenquimal(sarcomas): pred vía sanguinea. Metástasis. Factores quimiotácticos – Moléculas de adhesión.")
251
Model of Chemokine Regulation of Breast-Cancer Metastasis
Figure 1. Model of Chemokine Regulation of Breast-Cancer Metastasis. Metastasis is an orderly, multistep process involving the movement of cancer cells from the primary tumor to specific organs under the guidance of specific chemokines. First, cancerous mammary epithelial cells undergo clonal proliferation, invade local tissue, induce angiogenesis, and express CXC chemokine receptor 4 (CXCR4) on their surface. Then, cancer cells detach from the primary tumor, migrate across lymphatic and vascular walls in the tumor, and enter the systemic circulation. Cancer cells are arrested in vascular beds in organs that produce high levels of the CXCR4 ligand (CXCL12), which is expressed on the surface of vascular endothelial cells. Binding of CXCL12 to CXCR4 induces the migration of cancer cells into normal tissue, where the cells proliferate, induce angiogenesis, and form metastatic tumors. Breast-cancer cells do not usually metastasize to organs that produce low levels of CXCL12, such as the kidney. Murphy, P. M. N Engl J Med 2001;345:
 on their surface. Then, cancer cells detach from the primary tumor, migrate across lymphatic and vascular walls in the tumor, and enter the systemic circulation. Cancer cells are arrested in vascular beds in organs that produce high levels of the CXCR4 ligand (CXCL12), which is expressed on the surface of vascular endothelial cells. Binding of CXCL12 to CXCR4 induces the migration of cancer cells into normal tissue, where the cells proliferate, induce angiogenesis, and form metastatic tumors. Breast-cancer cells do not usually metastasize to organs that produce low levels of CXCL12, such as the kidney. Murphy, P. M. N Engl J Med 2001;345:")
252
Tumor Initiation and Metastasis
Figure 1. Tumor Initiation and Metastasis. The initiation and progression of tumors depend on the acquisition of specific functions by cancer cells at both the primary and metastatic sites. Functions associated with tumor initiation are provided by oncogenic mutations and inactivation of tumor-suppressor genes. Functions associated with the initiation of metastasis include functions to which tumor cells resort for local invasion and for circumventing hypoxia and other limitations facing a growing tumor. Most functions for the initiation of both the tumor and metastasis remain essential for cancer cells to continue their metastatic development. Functions for metastasis progression provide a local advantage in a primary tumor and a distinct and sometimes organ-specific function during metastasis. Cancer cells that are endowed with these three sets of functions still depend on functions associated with metastasis virulence; these functions confer a selective advantage solely during the adaptation and takeover of a specific organ microenvironment. Genes associated with each of these functions have been identified in recent years. Chiang A and Massague J. N Engl J Med 2008;359:

253
Patterns of Metastatic Spread of Solid Tumors.
Figure 2. Patterns of Metastatic Spread of Solid Tumors. Brain metastases may occur as a result of hematogenous spread late in the course of a widely metastatic tumor, or as a result of secondary metastasis from a primary or a metastatic tumor that can access the arterial circulation through the pulmonary venous circulation to seed the brain.38 Tumors with the highest incidence of brain metastases include lung carcinoma, breast carcinoma, melanoma, and to a lesser extent, renal-cell and colorectal carcinomas. Leptomeningeal disease may develop through the spread of cancer cells through perineural lymphatic vessels, and it is a sign of advanced disease. 38 Some tumors have a strong proclivity for dissemination to the lungs; for example, in one study, the rate of dissemination associated with sarcoma was 23%. 39 Other tumors that frequently spread to the lungs include renal-cell, colorectal, melanoma, and breast carcinomas. 40, 41 Gastrointestinal tumors easily access the liver circulation through the portal-vein system. The incidence of liver metastases is highest among patients with colorectal or pancreatic cancer, followed by breast and lung cancers. 40 Estrogen-receptor–negative breast-cancer tumors more often metastasize to visceral organs, including the liver, whereas estrogen-receptor–positive breast cancer more often metastasizes to the bone. 42 Bone metastasis occurs in patients with primary tumors associated with breast, lung, prostate, renal-cell, and colon cancer, in this order of frequency. 40 Bone metastases may be primarily osteolytic or osteoblastic, depending on the tumor of origin. Chiang AC, Massagué J. N Engl J Med 2008;359:

254
Genes, Functions, and Cellular Players in Organ-Specific Metastasis
Figure 3. Genes, Functions, and Cellular Players in Organ-Specific Metastasis. Organ-specific metastasis of breast-cancer cells involves different molecular players during colonization of the lungs and the bones. In the lung, cancer cells producing EREG, COX-2, MMP-1, and ANGPTL4 are better equipped to exit the pulmonary vasculature, since these factors alter the integrity of lung microcapillary endothelia; this function is less important for infiltration into the bone marrow because of the naturally fenestrated structure of the bone marrow sinusoid vasculature. In the lung parenchyma, the activity of the antidifferentiation gene ID1 and interactions with still unknown "niche" factors promote tumor reinitiation. In the bone marrow, stromal-cell-derived factor 1 (SDF-1), acting through its chemokine (C-X-C motif) receptor (CXCR4) on cancer cells, is thought to provide cell-survival functions. The secretion of parathyroid hormone-related peptide (PTHrP), interleukin-6, tumor necrosis factor{alpha} (TNF-{alpha}), interleukin-11, and other factors by cancer cells stimulates osteoblasts to release the ligand for the receptor activator of nuclear factor-{kappa}B (RANKL), which in turn stimulates osteoclast differentiation from myeloid progenitor cells. Other cancer cell-derived factors suppress the production of the RANKL antagonist osteoprotegerin, augmenting the efficacy of RANKL. The lytic action of osteoclasts releases bone matrix-associated growth factors, including transforming growth factor {beta} (TGF-{beta}), insulin-like growth factor I (IGF-I), and bone morphogenetic proteins (BMPs). IGF-I is a survival factor, and TGF-{beta} incites cancer cells to further release PTHrP, interleukin-11, and other prometastatic factors, establishing a vicious cycle. Chiang A and Massague J. N Engl J Med 2008;359:
, acting through its chemokine (C-X-C motif) receptor (CXCR4) on cancer cells, is thought to provide cell-survival functions. The secretion of parathyroid hormone-related peptide (PTHrP), interleukin-6, tumor necrosis factor{alpha} (TNF-{alpha}), interleukin-11, and other factors by cancer cells stimulates osteoblasts to release the ligand for the receptor activator of nuclear factor-{kappa}B (RANKL), which in turn stimulates osteoclast differentiation from myeloid progenitor cells. Other cancer cell-derived factors suppress the production of the RANKL antagonist osteoprotegerin, augmenting the efficacy of RANKL. The lytic action of osteoclasts releases bone matrix-associated growth factors, including transforming growth factor {beta} (TGF-{beta}), insulin-like growth factor I (IGF-I), and bone morphogenetic proteins (BMPs). IGF-I is a survival factor, and TGF-{beta} incites cancer cells to further release PTHrP, interleukin-11, and other prometastatic factors, establishing a vicious cycle. Chiang A and Massague J. N Engl J Med 2008;359:")
255
Defensa antitumoral INESPECÍFICA ESPECÍFICA Células natural killer
Macrófagos activados por citocinas producidas por linfocitos T Citocinas proinflamatorias (TNF alfa) ESPECÍFICA Linfocitos T citotóxicos (por Ags unidos a HLA I) Anticuerpos sintetizados por células B (citotoxicidad mediada por células dependiente de anticuerpos ADCC)
 ESPECÍFICA. Linfocitos T citotóxicos (por Ags unidos a HLA I) Anticuerpos sintetizados por células B (citotoxicidad mediada por células dependiente de anticuerpos ADCC)")
256
Antígenos tumorales A)Fallo de la célula tumoral en la reparación del DNA dañado y mutación (rojo) en proteína y presentación de péptido en HLA. B) Proteína normal puede ser sobreexpresada y sus péptidos presentados en superficie en cantidad excesiva. C) Modificación posttraslacional: proteína normal anormalmente procesada (spliced, glicosilada, fosforilada, lipidada) con péptidos anormales en la superficie de la célula.
Fallo de la célula tumoral en la reparación del DNA dañado y mutación (rojo) en proteína y presentación de péptido en HLA. B) Proteína normal puede ser sobreexpresada y sus péptidos presentados en superficie en cantidad excesiva. C) Modificación posttraslacional: proteína normal anormalmente procesada (spliced, glicosilada, fosforilada, lipidada) con péptidos anormales en la superficie de la célula.")
257
Tumor Antigens Eliciting T-Cell Immunity When Presented to Naive T Cells by Antigen-Presenting Dendritic Cells Figure 2. Tumor Antigens Eliciting T-Cell Immunity When Presented to Naive T Cells by Antigen-Presenting Dendritic Cells. Dendritic cells in the tumor or the tumor-draining lymph node take up dying tumor cells, tumor proteins, and tumor peptides and process and display them in their major-histocompatibility-complex (MHC) class I and class II molecules, as shown in Panel A. If properly activated by immunostimulatory tumor products or other factors in the tumor microenvironment, the dendritic cells induce effective tumor-specific CD4 and CD8 T cells. In Panel B, confocal microscopy shows dendritic cells (stained red with rhodamine phalloidin) that have taken up dying tumor cells (engineered to express green fluorescent protein). (Image courtesy of the University of Pittsburgh Cell and Molecular Imaging Facility.) Finn O. N Engl J Med 2008;358:
 class I and class II molecules, as shown in Panel A. If properly activated by immunostimulatory tumor products or other factors in the tumor microenvironment, the dendritic cells induce effective tumor-specific CD4 and CD8 T cells. In Panel B, confocal microscopy shows dendritic cells (stained red with rhodamine phalloidin) that have taken up dying tumor cells (engineered to express green fluorescent protein). (Image courtesy of the University of Pittsburgh Cell and Molecular Imaging Facility.) Finn O. N Engl J Med 2008;358:")
258
Tumor Antigens Eliciting T-Cell Immunity When Presented to Naive T Cells by Antigen-Presenting Dendritic Cells Figure 2. Tumor Antigens Eliciting T-Cell Immunity When Presented to Naive T Cells by Antigen-Presenting Dendritic Cells. Dendritic cells in the tumor or the tumor-draining lymph node take up dying tumor cells, tumor proteins, and tumor peptides and process and display them in their major-histocompatibility-complex (MHC) class I and class II molecules, as shown in Panel A. If properly activated by immunostimulatory tumor products or other factors in the tumor microenvironment, the dendritic cells induce effective tumor-specific CD4 and CD8 T cells. In Panel B, confocal microscopy shows dendritic cells (stained red with rhodamine phalloidin) that have taken up dying tumor cells (engineered to express green fluorescent protein). (Image courtesy of the University of Pittsburgh Cell and Molecular Imaging Facility.) Finn O. N Engl J Med 2008;358:
 class I and class II molecules, as shown in Panel A. If properly activated by immunostimulatory tumor products or other factors in the tumor microenvironment, the dendritic cells induce effective tumor-specific CD4 and CD8 T cells. In Panel B, confocal microscopy shows dendritic cells (stained red with rhodamine phalloidin) that have taken up dying tumor cells (engineered to express green fluorescent protein). (Image courtesy of the University of Pittsburgh Cell and Molecular Imaging Facility.) Finn O. N Engl J Med 2008;358:")
259
Immunostimulatory and Immunosuppressive Forces in the Tumor Microenvironment
Figure 3. Immunostimulatory and Immunosuppressive Forces in the Tumor Microenvironment. A growing tumor attracts many components of the host response. Tumor antigens and soluble tumor products attract dendritic cells to the tumor site. These dendritic cells take up tumor antigens, mature into interleukin-12-producing cells, and in the draining lymph node stimulate type 1 helper T-cell (Th1)-type CD4 T cells that produce interferon-{gamma}. These cells help expand the population of CD8 cytotoxic T-lymphocytes that can destroy tumor cells through effector molecules granzyme B and perforin. Another set of tumor antigens and soluble tumor products promote maturation of a different type of dendritic cell that makes proinflammatory cytokines interleukin-6 and tumor necrosis factor {alpha} (TNF-{alpha}) and give rise to type 2 helper T-cell (Th2)-type CD4 T cells that make interleukin-4 and interleukin-13 and are not effective in tumor rejection. This immunosuppressive environment also promotes generation of regulatory T cells and accumulation of macrophages and myeloid-derived suppressor cells (MDSC). At the time the tumor is diagnosed, the balance between the stimulatory and suppressive forces is in favor of tumor-induced suppression. Immunotherapy that targets the tumor with antibodies and T cells or augments antitumor Th1-type CD4 helper T cells and cytotoxic T lymphocytes with vaccines can tip the balance in favor of immunostimulation. GM-CSF denotes granulocyte-macrophage colony-stimulating factor, IDO indolamine-2,3-dioxygenase, iNOS inducible nitric oxide synthase, MCP-1 monocyte chemotactic protein 1, and TGF-{beta} transforming growth factor {beta}. Finn O. N Engl J Med 2008;358:
-type CD4 T cells that produce interferon-{gamma}. These cells help expand the population of CD8 cytotoxic T-lymphocytes that can destroy tumor cells through effector molecules granzyme B and perforin. Another set of tumor antigens and soluble tumor products promote maturation of a different type of dendritic cell that makes proinflammatory cytokines interleukin-6 and tumor necrosis factor {alpha} (TNF-{alpha}) and give rise to type 2 helper T-cell (Th2)-type CD4 T cells that make interleukin-4 and interleukin-13 and are not effective in tumor rejection. This immunosuppressive environment also promotes generation of regulatory T cells and accumulation of macrophages and myeloid-derived suppressor cells (MDSC). At the time the tumor is diagnosed, the balance between the stimulatory and suppressive forces is in favor of tumor-induced suppression. Immunotherapy that targets the tumor with antibodies and T cells or augments antitumor Th1-type CD4 helper T cells and cytotoxic T lymphocytes with vaccines can tip the balance in favor of immunostimulation. GM-CSF denotes granulocyte-macrophage colony-stimulating factor, IDO indolamine-2,3-dioxygenase, iNOS inducible nitric oxide synthase, MCP-1 monocyte chemotactic protein 1, and TGF-{beta} transforming growth factor {beta}. Finn O. N Engl J Med 2008;358:")
260
Manifestaciones del cáncer
Inespecíficas Astenia, anorexia, pérdida de peso (caquexia) Fiebre (linfomas…) Alteraciones biológicas (anemia, leucocitosis, reactantes fase aguda) Debidas a crecimiento e invasión tumoral. Tumoración Dolor (receptores nociceptivos) Trastorno funcional Estenosis víscera hueca Lesión mucosa (Ulceración- hemorragia- infección) Síndromes paraneoplásicos
 Fiebre (linfomas…) Alteraciones biológicas (anemia, leucocitosis, reactantes fase aguda) Debidas a crecimiento e invasión tumoral. Tumoración. Dolor (receptores nociceptivos) Trastorno funcional. Estenosis víscera hueca. Lesión mucosa (Ulceración- hemorragia- infección) Síndromes paraneoplásicos.")
261
SINDROMES PARANEOPLÁSICOS
No son consecuencia de su crecimiento, invasión ni secreciones hormonales propias del tejido de origen. Frecuentes. Endocrinos. ACTH por CPCpequeñas Metabólicos. Hipercalcema PTHrP Neurológicos. Eaton-Lambert Hematológicos. EPO (Policitemia) Procoagulantes (Tormboflebitis migrans) Dermatológicos. Dermatomiositis-Polimiositis
 Procoagulantes (Tormboflebitis migrans) Dermatológicos. Dermatomiositis-Polimiositis.")
262
Fisiopatología del Sistema Inmune
Remigio Cordero Torres 22 de Septiembre de 2015

263
Respuesta inmune patológica
Respuesta inmune inespecífica excesiva Síndrome respuesta inflamatoria sistémica Respuesta inmune específica excesiva Hipersensibilidad Autoinmunidad Defecto respuesta inmune inespecífica Defecto respuesta inmune específica Inmunodeficiencias Primarias Inmunodeficiencias Secundarias

264
Síndrome respuesta inflamatoria sistémica (SIRS)
Inflamación generalizada Citocinas proinflamatorias (TNF alfa) Causa infecciosa : SEPSIS Liberación endotoxinas Activación macrófagos sistémicos Causa no infecciosa: isquemia global, traumatismos graves Lesión y muerte celular Fagocitosis
 Causa infecciosa : SEPSIS. Liberación endotoxinas. Activación macrófagos sistémicos. Causa no infecciosa: isquemia global, traumatismos graves. Lesión y muerte celular Fagocitosis.")
265
Manifestaciones clínicas SIRS
Reacción de fase aguda Vasodilatación sistémica que puede llegar a producir shock Lesión y muerte tisular sistémica con desarrollo de: Síndrome de disfunción multiorgánica (MODS)
")
266
Síndrome respuesta inflamatoria sistémica (SIRS) Respuesta antiinflamatoria compensatoria (CARS) Síndrome de disfunción multiorgánica (MODS)
 Respuesta antiinflamatoria compensatoria (CARS) Síndrome de disfunción multiorgánica (MODS)")
267
Definición de SIRS clínico
Temperatura corporal > 38 o <36 Taquicardia (FC>90) Taquipnea (FR>20 resp/min) o hiperventilación (paCO2 <32) Leucocitosis > leuc/mm3 o formas inmaduras de serie blanca> 10%. Para establecer el diagnóstico se requieren al menos 2 criterios
 Taquipnea (FR>20 resp/min) o hiperventilación (paCO2 <32) Leucocitosis > leuc/mm3 o formas inmaduras de serie blanca> 10%. Para establecer el diagnóstico se requieren al menos 2 criterios.")
268
Respuesta inmune patológica
Respuesta inmune inespecífica excesiva Síndrome respuesta inflamatoria sistémica Respuesta inmune específica excesiva Hipersensibilidad Autoinmunidad Defecto respuesta inmune inespecífica Defecto respuesta inmune específica Inmunodeficiencias Primarias Inmunodeficiencias Secundarias

269
Hipersensibilidad La respuesta inmune específica dirigida contra un antígeno extraño se produce de forma excesiva o aberrante (reacción anormal cuantitativa o cualitativa). Sobre una predisposición genética actúa un factor ambiental que favorece una respuesta excesiva.
. Sobre una predisposición genética actúa un factor ambiental que favorece una respuesta excesiva.")
270
Autoinmunidad Respuesta inapropiada dirigida contra elementos estructurales del mismo individuo (autoantígenos) Pérdida de tolerancia inmunológica Factores genéticos: vía de presentación antigénica por moléculas HLA I y II. Alelos favorecedores de algunas enfermedades (HLA-B27 en Espondilitis Anquilosante). Factores ambientales
. Factores ambientales.")
271
Factores ambientales/autoinmunidad
Mimetismo molecular Coincidencia en estructura molecular de antígenos extraños y autoantígenos. (FR) Modificación de autoantígenos Cambio estructural en autoantígenos que se transforman en neoantígenos. (alfametildopa) Liberación de Antígenos de tejidos poco o nada relacionados con sistema inmune Lesión tisular (Uveítis autoinmune posttraumática)
 Modificación de autoantígenos. Cambio estructural en autoantígenos que se transforman en neoantígenos. (alfametildopa) Liberación de Antígenos de tejidos poco o nada relacionados con sistema inmune. Lesión tisular (Uveítis autoinmune posttraumática)")
272
Adapted from Kurtzke20 and Green and Patterson.21
The North-South Gradient in the Prevalence of Multiple Sclerosis (Panel A) and the Incidence of Type 1 Diabetes Mellitus (Panel B) in Europe Figure 2. The North-South Gradient in the Prevalence of Multiple Sclerosis (Panel A) and the Incidence of Type 1 Diabetes Mellitus (Panel B) in Europe. Adapted from Kurtzke20 and Green and Patterson.21 Bach, J.-F. N Engl J Med 2002;347:
 and the Incidence of Type 1 Diabetes Mellitus (Panel B) in Europe. Figure 2. The North-South Gradient in the Prevalence of Multiple Sclerosis (Panel A) and the Incidence of Type 1 Diabetes Mellitus (Panel B) in Europe. Adapted from Kurtzke20 and Green and Patterson.21. Bach, J.-F. N Engl J Med 2002;347:")
273
Inverse Relation between the Incidence of Prototypical Infectious Diseases (Panel A) and the Incidence of Immune Disorders (Panel B) from 1950 to 2000 Figure 1. Inverse Relation between the Incidence of Prototypical Infectious Diseases (Panel A) and the Incidence of Immune Disorders (Panel B) from 1950 to 2000. In Panel A, data concerning infectious diseases are derived from reports of the Centers for Disease Control and Prevention, except for the data on hepatitis A, which are derived from Joussemet et al.12 In Panel B, data on immune disorders are derived from Swarbrick et al.,10 Dubois et al.,13 Tuomilehto et al.,14 and Pugliatti et al.15 Bach, J.-F. N Engl J Med 2002;347:
 and the Incidence of Immune Disorders (Panel B) from 1950 to In Panel A, data concerning infectious diseases are derived from reports of the Centers for Disease Control and Prevention, except for the data on hepatitis A, which are derived from Joussemet et al.12 In Panel B, data on immune disorders are derived from Swarbrick et al.,10 Dubois et al.,13 Tuomilehto et al.,14 and Pugliatti et al.15. Bach, J.-F. N Engl J Med 2002;347:")
274
Reacciones inmunes que intervienen en hipersensibilidad y autoinmunidad
Reacción tipo I. Hipersensibilidad inmediata. Reacción tipo II. Citolítica o citotóxica Reacción tipo III. Inmunocomplejos Reacción tipo IV. Hipersensibilidad retardada

275
Four Types of Hypersensitivity Reactions.
Patterson R et al. N Engl J Med 1976;295:

277
Reacción tipo I. Hipersensibilidad inmediata
El Antígeno se denomina alérgeno Los Anticuerpos (IgE) se denominan reaginas Predisposición genética atopia Implicada en enfermedades alérgicas Puede tener repercusión sistémica: anafilaxia
 se denominan reaginas. Predisposición genética atopia. Implicada en enfermedades alérgicas. Puede tener repercusión sistémica: anafilaxia.")
278
Reacción tipo I. Hipersensibilidad inmediata
Acceso de alérgeno: Inhalación, Ingestión; Inoculación; Contacto con piel o mucosas. Primer contacto Captación por células dendríticas y linfocitos B Presentación junto HLAII a linfocitos T cooperadores que secretan citocinas con patrón Th2 (IL-4 /IL-5) IL-4 estimula secreción IgE específica por LinfsB: unión a mastocitos o basófilos. IL-5 estimula granulopoyesis eosinofílica Nuevos contactos
 IL-4 estimula secreción IgE específica por LinfsB: unión a mastocitos o basófilos. IL-5 estimula granulopoyesis eosinofílica. Nuevos contactos.")
279
Reacción tipo I. Hipersensibilidad inmediata
Acceso de alérgeno: Inhalación, Ingestión; Inoculación; Contacto con piel o mucosas. Primer contacto Nuevos contactos Unión a IgE en superficie de mastocitos o basófilos Exocitosis mediadores: histamina Síntesis y liberación de factor activador de plaquetas Síntesis y secreción de citocinas proinflamatorias (IL-1, TNF alfa) IL-4, IL-5, leucotrienos
 IL-4, IL-5, leucotrienos.")
280
Reacción tipo I a) Primer contacto con alérgeno b) Nuevo contacto con alérgeno
 Primer contacto con alérgeno b) Nuevo contacto con alérgeno")
282
Reacción tipo I. Hipers inmediata-Manifestaciones clínicas
Rinitis Alérgica. Fiebre del heno Asma bronquial. Hiperreactividad bronquial Dermatitis atópica Urticaria Angioedema Eccema atópico Anafilaxia Shock anafiláctico: expresión más grave

283
Reacción tipo II. Citolítica Citotóxica Citoestimulante Citoinhibidora

284
Reacción tipo II. Citolítica
Ags membrana cél sanguineas (o depositado en ella). Anticuerpos IgM o IgG 1,2,3 Activación del Complemento Completa: Lisis intravascular . (IgM) Formación de complejo ataque membrana (MAC) Incompleta: Lisis extravascular (IgG) Frag C3b une membrcelular y macrófago esplénico Acción Opsonizante de IgG. Unión a membrana de hematíe y a receptor Fc macrófago esplénico. Anemia hemolítica, neutropenia, plaquetopenia
. Anticuerpos IgM o IgG 1,2,3. Activación del Complemento. Completa: Lisis intravascular . (IgM) Formación de complejo ataque membrana (MAC) Incompleta: Lisis extravascular (IgG) Frag C3b une membrcelular y macrófago esplénico. Acción Opsonizante de IgG. Unión a membrana de hematíe y a receptor Fc macrófago esplénico. Anemia hemolítica, neutropenia, plaquetopenia.")
285
Reacción tipo II citolítica

286
Reacción tipo II. Citotóxica
Ags localizados en tejidos (Autoantígenos) Anticuerpos IgM o IgG 1,2,3 Activación del Complemento Vía clásica: C3a, C4a, C5a (anafilotoxinas) Liberación de histamina : vasodilatación y aumento de permeabilidad Atracción de Neutrófilos (C5a) Activación de Neutrófilos y Macrófagos Fragmento Fc IgG Síndrome de Goodpasture (lesión alveolar y glomerular)
 Anticuerpos IgM o IgG 1,2,3. Activación del Complemento. Vía clásica: C3a, C4a, C5a (anafilotoxinas) Liberación de histamina : vasodilatación y aumento de permeabilidad. Atracción de Neutrófilos (C5a) Activación de Neutrófilos y Macrófagos. Fragmento Fc IgG. Síndrome de Goodpasture (lesión alveolar y glomerular)")
287
Mecanismos lesionales en reacción II citotóxica y reacción III

288
Reacción tipo II. Citoestimulante o citoinhibidora
Antígenos: receptores de membrana Producen alteración funcional sin lesión. Reacción Citoestimulante Antígenos: receptores de TSH Acs: Igs estimulantes tiroides (TSI) Enfermedad deGraves – Basedow. Reacción Citoinhibidora Antígenos: receptor Acetilcolina sarcolema Miastenia gravis.
 Enfermedad deGraves – Basedow. Reacción Citoinhibidora. Antígenos: receptor Acetilcolina sarcolema. Miastenia gravis.")
289
Pathogenesis of Graves' Disease
Figure 2. Pathogenesis of Graves' Disease. Excess production of thyroid hormone is caused by the activation of thyrotropin receptors by thyroid-stimulating antibodies produced within and outside the thyroid gland. The intrathyroidal inflammatory cells also produce cytokines, such as interleukin-1, tumor necrosis factor {alpha}, and interferon-{gamma}, that induce the expression of adhesion molecules such as CD54, regulatory molecules such as CD40, and HLA class II molecules, which in turn activate local inflammatory cells. These cytokines also induce thyroid cells to synthesize cytokines that may help sustain the intrathyroidal autoimmune process. Antithyroid drugs reduce the production of thyroidal cytokines -- an ability that may explain their immunomodulatory effects (which include a decrease in the production of thyroid-stimulating antibody) -- contributing to remission in some patients. Pathogenesis of Graves' Disease Weetman, A. P. N Engl J Med 2000;343:
 -- contributing to remission in some patients. Pathogenesis of Graves Disease. Weetman, A. P. N Engl J Med 2000;343:")
290
Pathogenesis of Graves' Disease
Figure 2. Pathogenesis of Graves' Disease. Excess production of thyroid hormone is caused by the activation of thyrotropin receptors by thyroid-stimulating antibodies produced within and outside the thyroid gland. The intrathyroidal inflammatory cells also produce cytokines, such as interleukin-1, tumor necrosis factor {alpha}, and interferon-{gamma}, that induce the expression of adhesion molecules such as CD54, regulatory molecules such as CD40, and HLA class II molecules, which in turn activate local inflammatory cells. These cytokines also induce thyroid cells to synthesize cytokines that may help sustain the intrathyroidal autoimmune process. Antithyroid drugs reduce the production of thyroidal cytokines -- an ability that may explain their immunomodulatory effects (which include a decrease in the production of thyroid-stimulating antibody) -- contributing to remission in some patients. Pathogenesis of Graves' Disease Weetman, A. P. N Engl J Med 2000;343:
 -- contributing to remission in some patients. Pathogenesis of Graves Disease. Weetman, A. P. N Engl J Med 2000;343:")
291
Reacción tipo III. Reacción por Inmunocomplejos
Inmunocomplejos circulantes Antígenos exógenos : fármacos, proteínas, virus Antígenos endógenos: células destruidas, tumores Depósito dependiente de: tamaño, capacidad de eliminación, propiedades físicoquímicas, determinates anatómicos y hemodinámicos Reacción citotóxica semejante a tipo II Vasculitis , Lupus eritematoso sistémico. Inmunocomplejos formados in situ

292
Reacción tipo III. Reacción por Inmunocomplejos
Inmunocomplejos circulantes Inmunocomplejos formados in situ Formación: Antígenos atrapados y depositados en tejido Glomerulonefritis Exposición repetida al antígeno inoculado o inhalado Alveolitis alérgica extrínseca Activación de reacción citotóxica II Activación del complemento Activación de macrófagos y neutrófilos

293
Mecanismos lesionales en reacción II citotóxica y reacción III

294
Reacción tipo IV. Hipersensibilidad retardada
Mediada por linfocitos T colaboradores Bacterias resistentes en macrófagos Antígenos expresados en membrana Activación linfocitos T colaboradores Secreción de IL-2 e IFN gamma (Th1) Activación de Macrófago Secreción IL-12 – Activación de macrófago Mycobacterium tuberculosis Dermatitis de contacto Otras granulomatosis: sarcoidosis Mediada por linfocitos T citotóxicos
 Activación de Macrófago. Secreción IL-12 – Activación de macrófago. Mycobacterium tuberculosis. Dermatitis de contacto. Otras granulomatosis: sarcoidosis. Mediada por linfocitos T citotóxicos.")
295
Respuesta inmune patológica
Respuesta inmune inespecífica excesiva Respuesta inmune específica excesiva Defecto respuesta inmune inespecífica Alteraciones funcionales de fagocitos Déficiencias del sistema del complemento Defecto respuesta inmune específica Inmunodeficiencias Primarias Inmunodeficiencias Secundarias

296
Inmunodeficiencias primarias A) combinadas B) alteración respuesta linfoide B
 combinadas B) alteración respuesta linfoide B")
Presentaciones similares
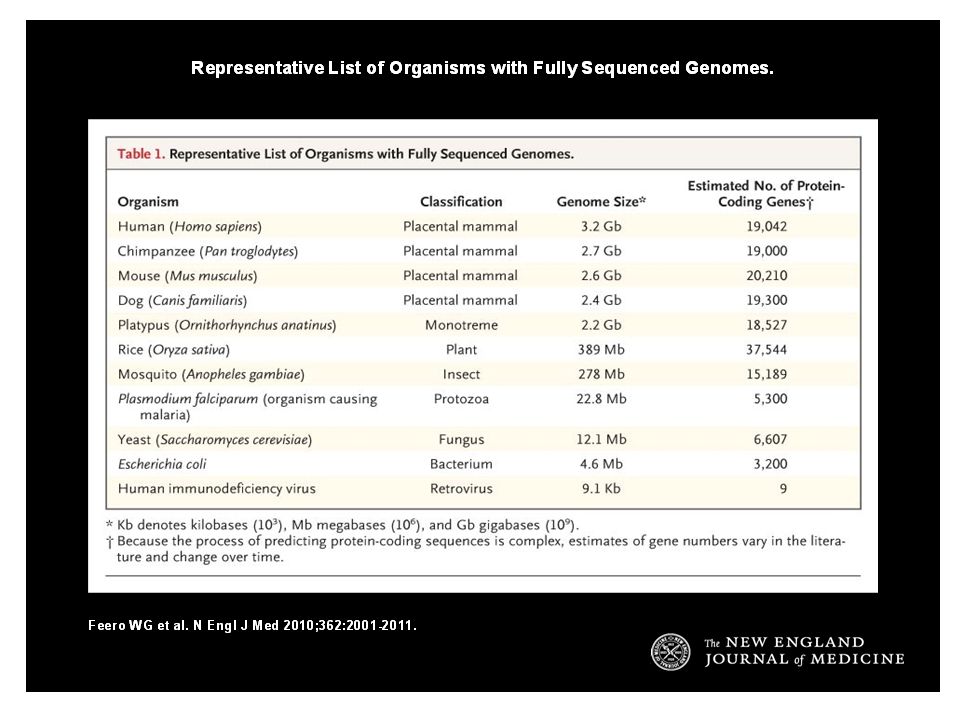


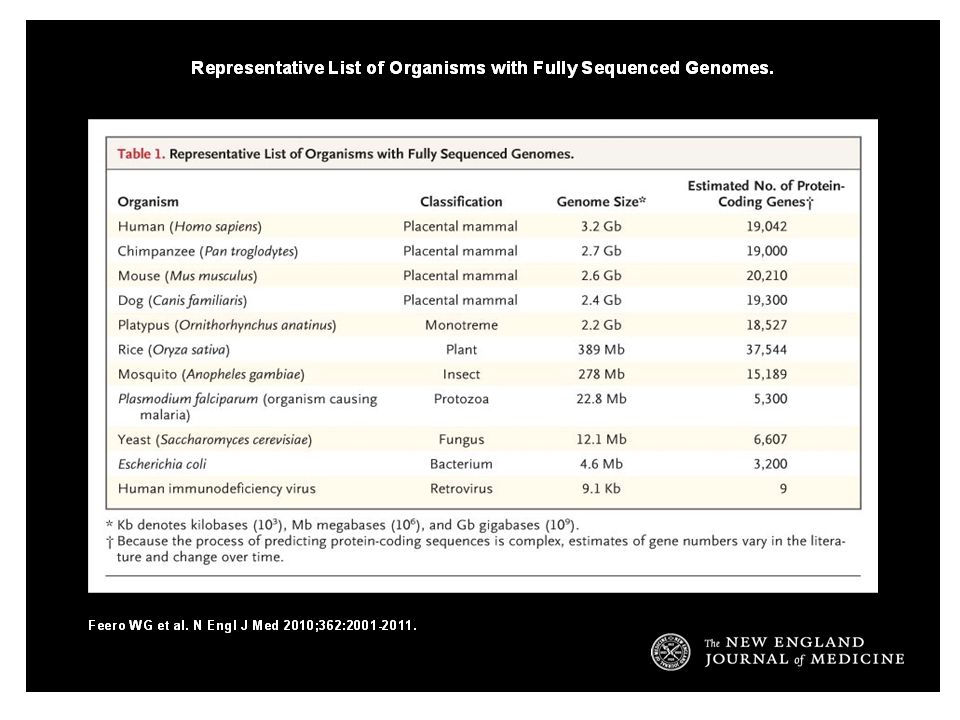

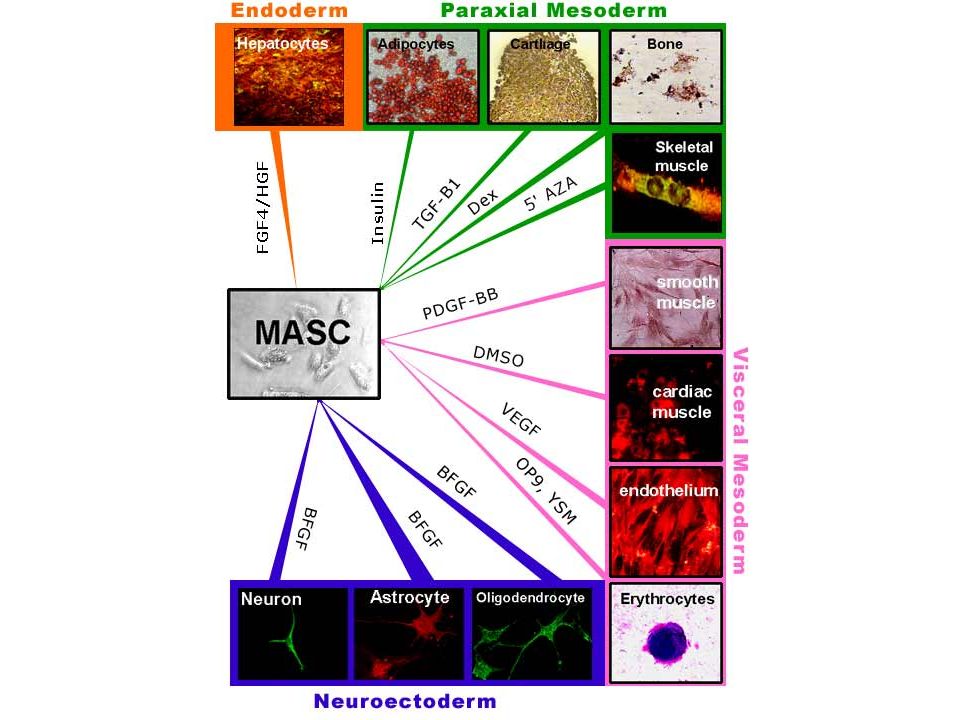




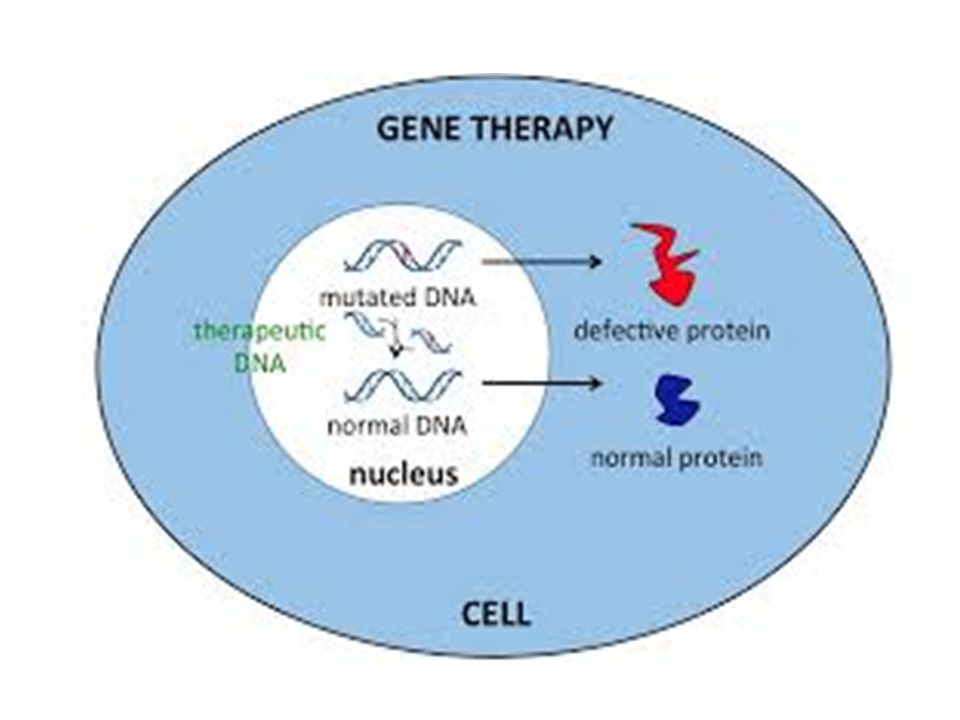
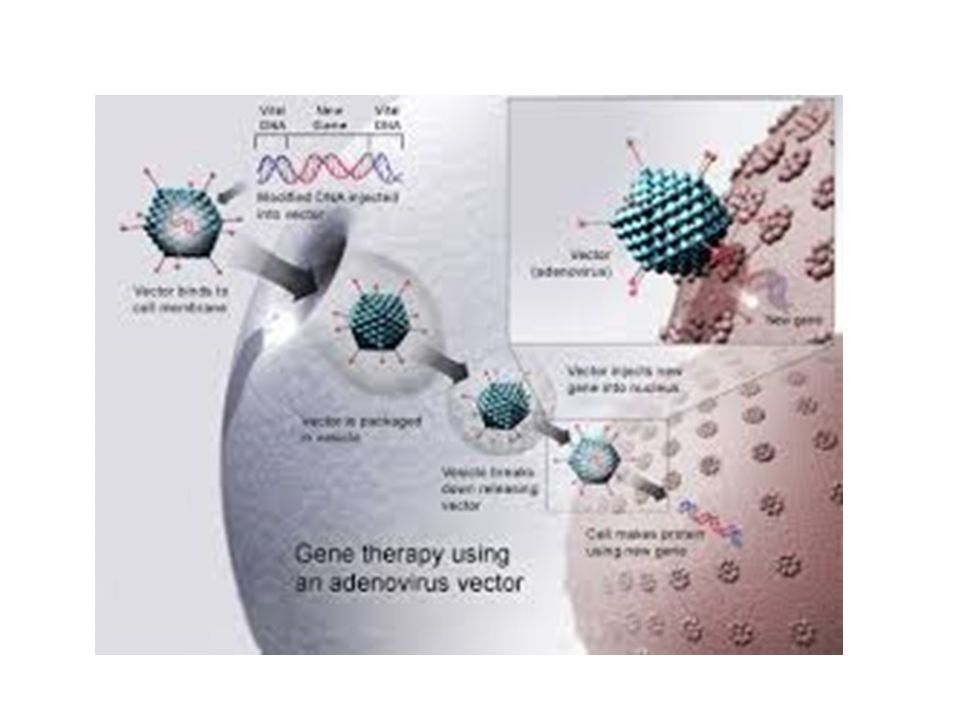












 ESPECÍFICAS (Respuesta inmunitaria) – La unión antígeno anticuerpo es específica.>")






