Descargar la presentación
La descarga está en progreso. Por favor, espere

1
HIPERPLASIA SUPRARRENAL CONGÉNITA
Cecilia Narvaez 2013

2
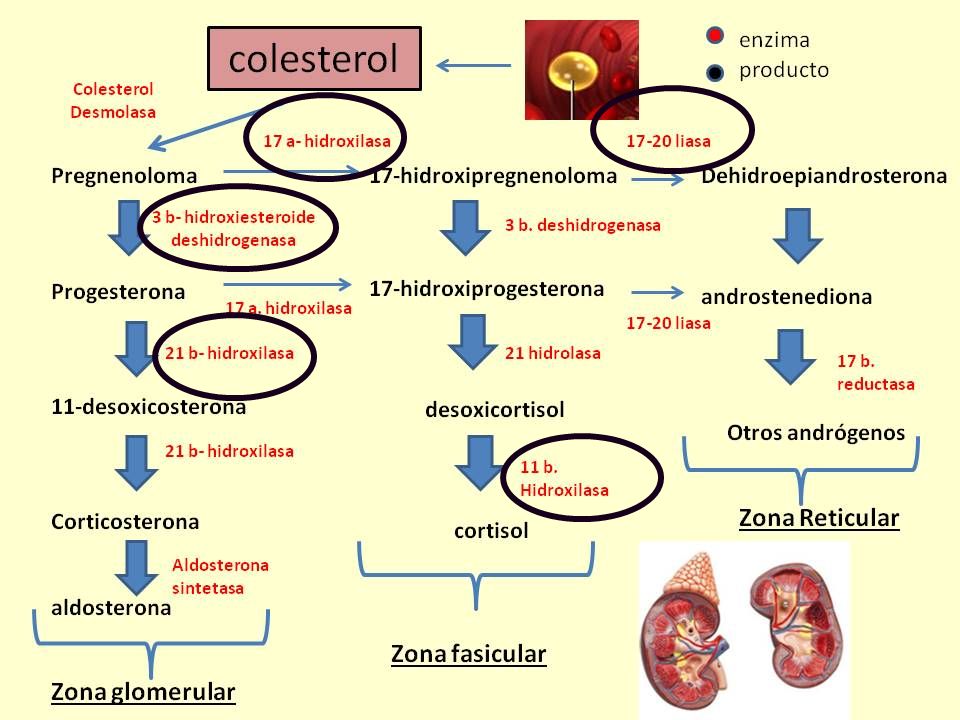
Engloba un grupo de enfermedades hereditarias autosómicas recesivas Déficit de enzimas que intervienen en la transformación del COLESTEROL Déficit en la biosíntesis del CORTISOL

4
Por orden de frecuencia:
21-α-hidroxilasa > 90% 11-β-hidroxilasa 3-β-hidroxiesteroide deshidrogenasa 17-α-hidroxilasa/17-20 desmolasa

5
HSC 21-α-hidroxilasa 11-β-hidroxilasa
Afectación de la biosíntesis suprarrenal Afectación de biosíntesis De hormonas sexuales, Cortisol y aldosterona 21-α-hidroxilasa 11-β-hidroxilasa 3-β-hidroxiesteroide deshidrogenasa 17-α-hidroxilasa/ 17-20 desmolasa

6
HIPOTÁLAMO secreta corticotropina (CRF) liberando ACTH
actúa ADENOHIPÓFISIS liberando ACTH controla la esteroidogénesis en la glándula suprarrenal; ya que, estimula las diferentes enzimas implicadas en la síntesis de cortisol, S- DHEA y aldosterona.

7
síntesis del cortisol secreción de ACTH.
CORTISOL regula la secreción de CRF y ACTH El bloqueo en la acción de alguna de las enzimas produce : síntesis del cortisol secreción de ACTH. HIPERTROFIA DE LA GL. RENAL

8
HSC QUE AFECTA UNICAMENTE A LA BIOSINTESIS SUPRARRENAL
Principal causa de virilización anormal en el Sexo FEMENINO Causa del 90-95% de los casos de PHF

9
Déficit de 21-Hidroxilasa
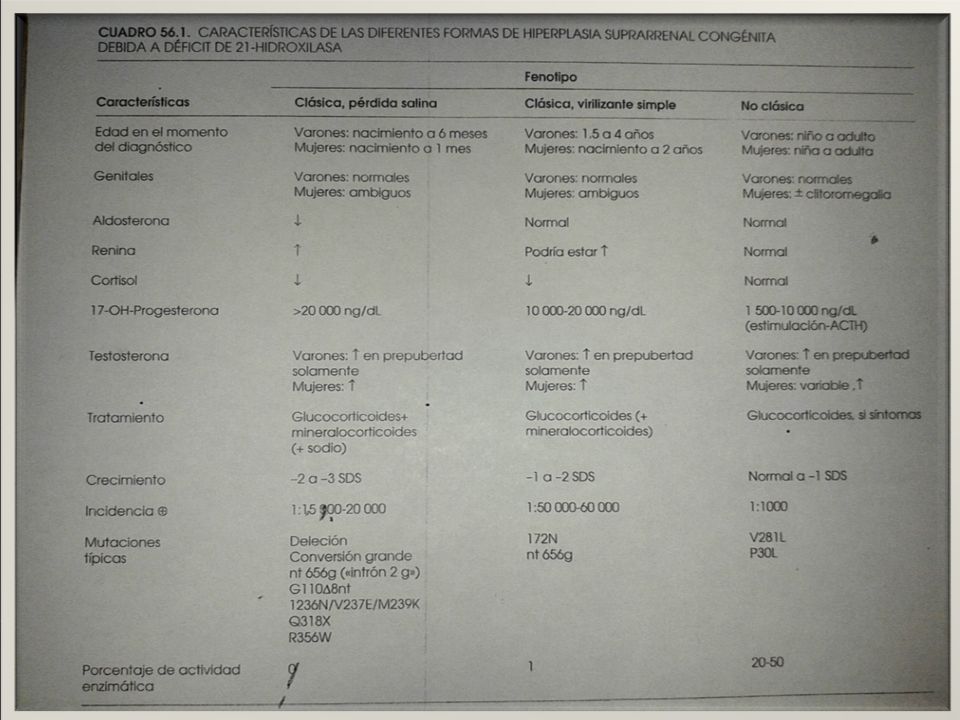
Forma más frecuente de HSC >90% - Incidencia de forma clásica 1/ nacidos vivos. - Incidencia de portadores de la mutación de 1 / 60 individuos (heterocigotos). - Variación en diferentes áreas geográficas y grupos étnicos. esquimales Yupic de Alaska (1 / 280) Brasil (1 / 7.500) y Filipinas (1 / 7.000). - EE.UU, la incidencia es mayor en población blanca que en la afroamericana EPIDEMIOLOGIA
. - Variación en diferentes áreas geográficas y grupos étnicos. esquimales Yupic de Alaska (1 / 280) Brasil (1 / 7.500) y Filipinas (1 / 7.000). - EE.UU, la incidencia es mayor en población blanca que en la afroamericana. EPIDEMIOLOGIA.")
10
Insuficiencia suprarrenal Pérdida salina Hiper- androgenismo
DEFICIT 21-HIDROXILASA Insuficiencia suprarrenal Pérdida salina Hiper- androgenismo

11
21-hidroxilasa regula FISIOPATOLOGIA progesterona a
hidroxilación de progesterona a 11desoxicorticosterona precursor de aldosterona hidroxilación de17-hidroxiprogesterona en desoxicortisol precursor de cortisol LA DISMINUCION DE CORTISOL ACTH Y ESTA SE DESVIARA HACIA LA BIOSINTESIS DE ANDROGENOS

12
La podemos dividir en dos grandes tipos:
21-α-hidroxilasa La forma de presentación clínica varía dependiendo del grado de déficit enzimático. La podemos dividir en dos grandes tipos:

13
Virilizante simple CLÁSICA : inicio intra útero
21-α-hidroxilasa CLÁSICA : inicio intra útero Síndrome perdedor de sal Virilizante simple NO CLÁSICA O TARDÍA

14
- mutación produce un déficit total de cortisol y aldosterona
21-α-hidroxilasa FORMA CLASICA CON PERDIDA SALINA - El 75% de los pacientes. - mutación produce un déficit total de cortisol y aldosterona

15
produce una excreción excesiva de Na por la orina y
21-α-hidroxilasa Deficit de ALDOSTERONA Deficit de CORTISOL produce una excreción excesiva de Na por la orina y la eliminación urinaria de K Hiponatremia Hiperpotasemia Eliminación de Na arrastra agua y Hco3 Hipovolemia, Hipotensión Acidosis metabólica . Produce del tono vascular, del inotropismo cardíaco e hipoglucemia. Por su actividad mineralocorticoide, contribuye a la Hiponatremia, Deshidratación e Hipotensión.

16
Se manifiesta entre el 5º-15º días de vida
- vómitos - escasa ganancia ponderal - diarrea - hipotensión- shock y colapso cardiovascular alteraciones bioquímicas: - hipoglucemia - hiponatremia <120 mEq /l - hiperpotasemia > 10 mEq/l -acidosis metabólica. Ph 7.1

17
En el feto con HSC la placenta y riñón y glándula suprarrenal Materna, permiten mantener al feto una homeostasis electrolítica. Por ello el S° de pérdida salina no se desarrolla hasta después del nacimiento. Por lo general durante la 2° semana de vida

18
El de los andrógenos desde la semana 7ª de gestación produce:
21-α-hidroxilasa El de los andrógenos desde la semana 7ª de gestación produce: un trastorno de la diferenciación sexual: - aumento del tamaño del clítoris - fusión parcial o completa de los labios mayores - vagina en apertura común con la uretra (seno urogenital) - El útero, ovarios y trompas de Falopio tendrán un desarrollo normal.
 - El útero, ovarios y trompas de Falopio tendrán un desarrollo normal.")
19
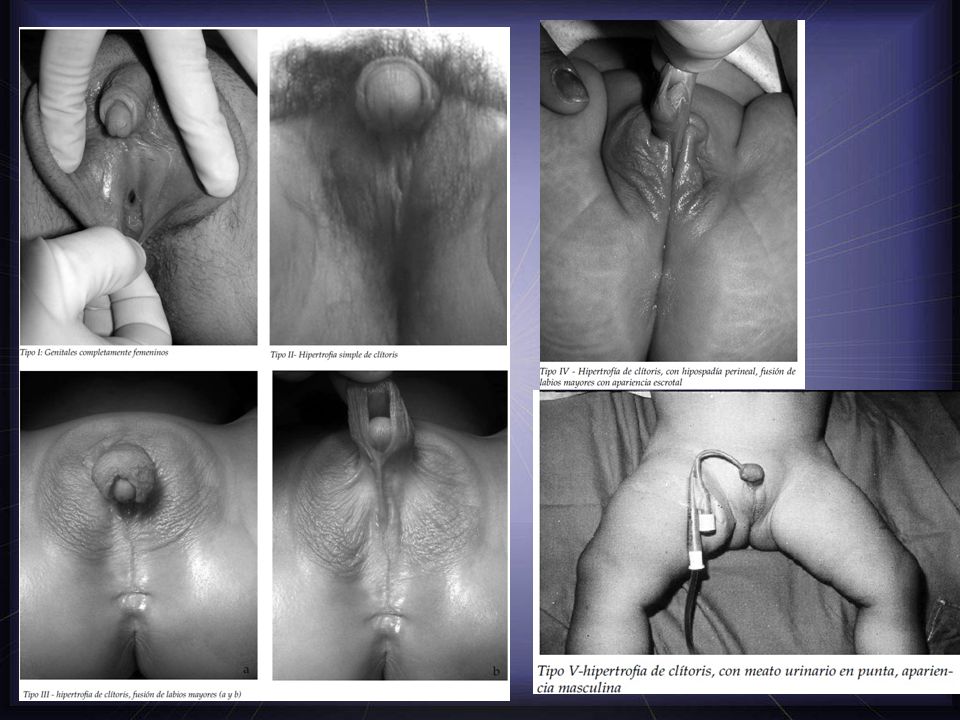
Tipo I Genitales completamente femeninos.
El examen físico es el primer elemento de orientación. Utilizamos los 5 Tipos de PRADER: Tipo I Genitales completamente femeninos. Tipo II Hipertrofia simple de clítoris. Tipo III hipertrofia de clítoris, fusión de labios mayores. Tipo IV Hipertrofia de clítoris, con hipospadía perineal, fusión de labios mayores con apariencia escrotal. Tipo V hipertrofia de clítoris, con meato urinario en punta, apariencia masculina

21
-Pérdidas salinas -Hiperpigmentación de genitales externos -Aumento del tamaño del pene

23
Forma virilizante simple:
21-α-hidroxilasa Forma virilizante simple: - 25% de los casos. hay un mínimo de actividad enzimática residual que determina que la síntesis de aldosterona y de cortisol no estén totalmente suprimidas, por lo que no presentan crisis de pérdida salina, a pesar de que los niveles de renina están elevados

24
trastorno de la diferenciación sexual en el momento del nacimiento.
Las niñas son identificadas precozmente por la virilización de los genitales externos signos de virilización desapercibidos manifestaciones clínicas tardía: aparición precoz de vello púbico, axilar pene alargado, escroto pigmentado aceleración de la velocidad de crecimiento y de la maduración ósea.

25
Forma no clásica o tardía
suficiente act. enzimática que produce adecuadamente cortisol y aldosterona y la producción de andrógenos no es excesiva. incidencia de 1/ 500 recién nacidos vivos. +++ judíos del este europeo. recién nacidos asintomáticos, apareciendo los síntomas con posterioridad. A veces, el diagnóstico se realiza en la etapa adulta por problemas de infertilidad. un grupo de pacientes que permanecen asintomáticos. 21-α-hidroxilasa

26
Forma no clásica o tardía
21-α-hidroxilasa Forma no clásica o tardía adrenarquia prematura acné aceleración del crecimiento, con edad ósea avanzada. - infancia: adrenarquia prematura acné aceleración del crecimiento y de la edad ósea. - adolescencia: hirsutismo irregularidades menstruales amenorrea.

27
Diagnóstico Genético CLINICO BIOLOGICO GENETICO Del déficit de 21-OH
El gen responsable del déficit de 21-OH se denomina CYP21A2, se localiza en el brazo corto del cromosoma 6p21.3, en la región III del sistema HLA. CLINICO BIOLOGICO GENETICO

28
Diagnóstico Biológico
Cifras plasmáticas basales de 17-OHP >150 a 1500nmol O >5000 a ng/dL Las demás H Esteroideas NO ayudan al diagnóstico Cifras de ACTH plasmáticas y de ARP (actividad de renina plasmática) están muy elevadas en las formas clásicas El Test de estimulación corto con ACTH mide directamente la integridad funcional de las glándulas suprarrenales e indirectamente la función del eje hipotálamo-hipofisario
 están muy elevadas en las formas clásicas. El Test de estimulación corto con ACTH. mide directamente la integridad funcional de las glándulas suprarrenales e indirectamente la función del eje hipotálamo-hipofisario.")
29
SCREENING NEONATAL DEL DÉFICIT DE 21-HIDROXILASA
objetivos: a) anticiparse a la aparición de una crisis de pérdida salina grave y potencialmente letal. b) evitar una incorrecta asignación de sexo en una niña con genitales externos virilizados c) diagnosticar precozmente las formas virilizantes simples para evitar la hiperandrogenización durante la infancia que determinará una talla final baja
 anticiparse a la aparición de una crisis de pérdida salina grave y potencialmente letal. b) evitar una incorrecta asignación de sexo en una niña con genitales externos virilizados. c) diagnosticar precozmente las formas virilizantes simples para evitar la hiperandrogenización durante. la infancia que determinará una talla final baja.")
30
Screening Neonatal determinación de 17-OHP en sangre total en papel de filtro + Medir 17-OHP en suero realizar finalmente un diagnóstico genético.

31
- Resultados: r.n de término - sospecha : + (10 ng/mL)
21-α-hidroxilasa Screening neonatal - Se realiza 17-hidroxiprogesterona (17-OHP) en sangre seca, por punción del talón en tarjetas de papel. - Resultados: r.n de término - sospecha : + (10 ng/mL) - patológicos : + (20 ng/mL) determinar 17-OHP sérica - entre ng/mL, repetir la muestra
 en sangre seca, por punción del talón en tarjetas de papel. - Resultados: r.n de término. - sospecha : + (10 ng/mL) - patológicos : + (20 ng/mL) determinar. 17-OHP sérica. - entre ng/mL, repetir la muestra.")
32
Falsos Positivos Falsos Negativos. Los prematuros y los neonatos
con enfermedades concomitantes, por estar sometidos a un estrés adicional cuando la muestra de sangre total se obtiene en las primeras 24 horas de vida Falsos Negativos. se han descrito, pero muy raramente, falsos negativos en casos de formas virilizantes simples que motivaron un retraso en el diagnóstico

33
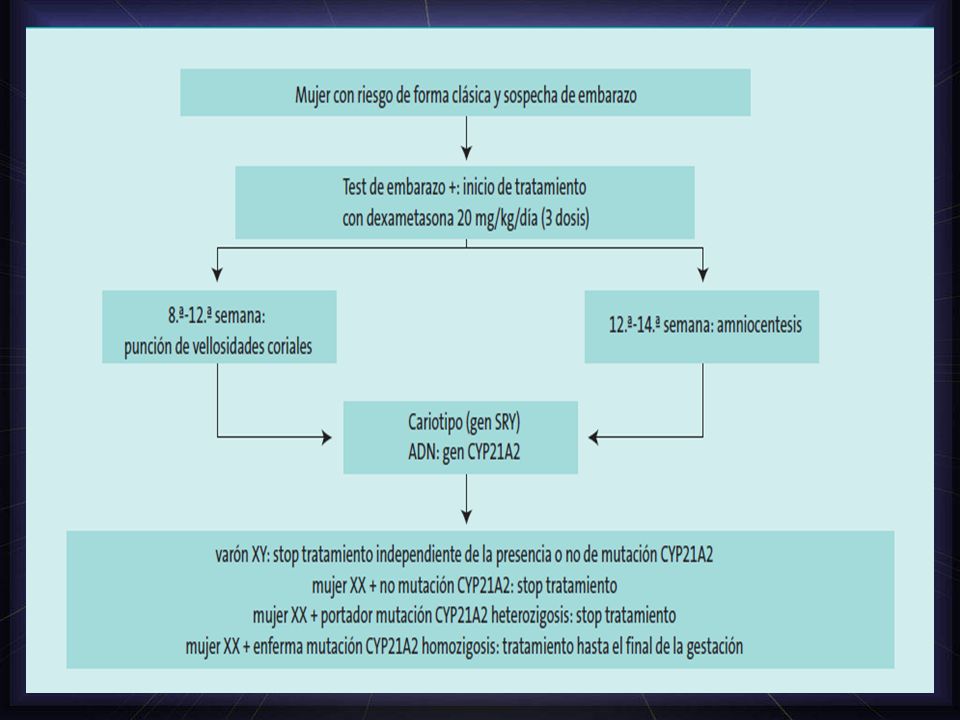
21-α-hidroxilasa Diagnóstico prenatal - Se aconseja en los casos en los que los padres tienen un hijo previo afecto o si son portadores de mutaciones. - 1er trimestre: ADN por biopsia de vellosidades coriónicas - 2do trimestre: amniocentesis. - Se analiza el gen CYP21 y se buscan las mutaciones más habituales. - administra a la madre dexametasona hasta el final del embarazo.

35
Déficit de 11- -hidroxilasa
5-8% de los casos incidencia entre1/ y 1/ +++ judíos israelíes de origen norteafricano. hidroxila : 11-desoxicortisol a cortisol desoxicorticosterona a cortisona de la síntesis de aldosterona y cortisol, que produce un de ACTH y, 2 rio de los precursores androgénicos El gen: región Q21-22 del brazo largo del cromosoma 8 y se denomina CYP11B1.

36
favorece la reabsorción renal de Na y la eliminación de k.
11 -hidroxilasa NO presentan signos - síntomas de insuf. suprarrenal, porque de desoxicorticosterona que tiene actividad mineralcorticoide favorece la reabsorción renal de Na y la eliminación de k. Hipertensión arterial e Hipopotasemia

37
de precursores androgénicos
11-hidroxilasa de precursores androgénicos virilización excesiva en las niñas, genitales ambiguos en el momento del parto. niños: produce hiperpigmentación local y alargamiento del pene, puede pasar desapercibido y manifestarse con posterioridad Diagnóstico : determinar niveles de 11-desoxicortisol Niveles de renina plasmática suprimidos El diagnóstico es neonatal

38
Déficit de 3- hidroxiesteroidedeshidrogenasa
Menos del 2% El déficit produce en la síntesis de cortisol, aldosterona y androstendiona, con aumento en la secreción de DHEA. El gen: región p13 del cromosoma 1. Es la única que produce trastorno de la diferenciación sexual en niños y niñas deficiencia de androstendiona síntesis de testosterona y ausencia de virilización de 17-hidroxipregnenolona y DHEA, 17-hidroxiprogesterona,

39
Déficit de 17-hidroxilasa/17-20-liasa
Menos del 1% El gen: en el cromosoma 10 y se denomina CYP17. NO sintetiza Cortisol pero SI desoxicorticosterona en exceso Hipertensión arterial Hipopotasemia supresión de la secreción de renina y aldosterona. Niños: fenotipo femenino o ambigüedad sexual. Niñas: adolescencia :retraso puberal, amenorrea primaria e HTA progesterona y desoxicorticosterona

40
Hiperplasia lipoidea congénita
Es la forma más rara - de 100 pacientes, mayoría japoneses. grave alteración de la esteroidogénesis, con gran acúmulo de colesterol, por un déficit en la proteína STAR. El gen: cromosoma 8. insuf. suprarrenal aguda en los 1ros días de vida (hipoglucemia, hipotensión,hiponatremia, shock) todos los precursores suprarrenales están Los recién nacidos varones tienen genitales ambiguos o de aspecto femenino, niñas genitales externos femeninos normales.
 todos los precursores suprarrenales están. Los recién nacidos varones tienen genitales ambiguos o de aspecto femenino, niñas genitales externos femeninos normales.")
41
TRATAMIENTO Insuficiencia suprarrenal aguda - expanción SF 20 mL/kg.
- Hiponatremia: < 120 mEq/L: ClNa 3% a 2-3 mL/kg en minutos. mEq/L : corregir como hiponatremia grave > 125 mEq/L: corrección lenta -Hipoglucemia sintomática: Dx 25%: 2-4 mL/kg. - Hiperpotasemia > 7 mEq/L y/o signos ECG: Gluconato cálcico 10%: 0,5 mL/kg iv en minutos. Bicarbonato sódico 1M: 1 mEq/kg a pasar en 30 min. - El tratamiento se sustenta en la terapia susitutiva con corticoides y mineralcorticoides.

42
TRATAMIENTO Tratamiento de mantenimiento que se asocian síndrome pierde sal - Déficit de cortisol : glucocorticoides indefinidamente, para inhibir la secreción de CRH. - Finalidad : administrar la mínima dosis que suprima la producción de andrógenos sin alterar la vel. de crecimiento ni la ganancia de peso. - Situaciones intercurrentes ej: infecciones triplicar la dosis de mantenimiento

43
hidrocortisona entorno a 15mg/m2/día a veces más, varía en función de la edad y estadío puberal del paciente pubertad 20 mg/m2/día neonatos 5 mg/día dividido en tres dosis, que supone aproximadamente una dosis de 25 mg/m2/día; las dosis suprafisiológicas administradas en el neonato son necesarias para suprimir adecuadamente los andrógenos suprarrenales y minimizar la posibilidad de desarrollar una insuficiencia suprarrenal.

44
Mineralocorticoides:
9-alfa-fludrocortisona 0,05-0,2 mg/día divididos en 3 dosis Cloruro sódico µg/m2/día durante el 1er año de vida.

45
TRATAMIENTO Tratamiento de mantenimiento de las forma virilizante:
tratamiento con glucocorticoides a dosis inferiores que el síndrome pierde sal. Forma no clásica : tratar sólo aquellos que presenten aceleración del crecimiento, con de la edad ósea y empeoramiento del pronóstico de talla Otros déficit : tratamiento con glucocorticoides

46
bibliografía INSUFICIENCIA SUPRARRENAL I Rica, G Grau, A Vela Servicio de Endocrinología Infantil. Hospital de Cruces. Barakaldo (Bizkaia) TRATADO DE ENDOCRINOLOGIA PEDIATRICA Autor: POMBO. M.HIPERPLASIA SUPRARRENAL CONGÉNITA JI Labarta Aizpún, A de Arriba Muñoz, Á Ferrández Longás Unidad de Endocrinología. Servicio de Pediatría. Hospital Infantil Miguel Servet. Zaragoza.

Presentaciones similares